


DIESES REFERAT HAT SICH DURCH DIE ENORME KOMPLEXITÄT DES THEMAS ZU EINEM BUCH ENTWICKELT. WENN SIE DIE NEUESTE VERSION ERHALTEN MÖCHTEN, DANN KONTAKTIEREN SIE MICH BITTE ÜBER DAS KONTAKTFORMULAR.
Vorwort
Um es gleich vorwegzunehmen: Eine Krankheit mit dem Namen Amyotrophe Lateralsklerose existiert nicht. Punkt, Pause, sacken lassen und entspannen...und noch mehr entspannen...Für Amyotrophe Lateralsklerose gibt es kein Diagnoseverfahren und daher ist dieser Name nur eine Art Platzhalter für Menschen die unter einem unerkannten Syndrom leiden, bei dem unter anderem motorische Nerven belastet werden und unter gewissen Umständen degenerieren können, aber nicht müssen. Nochmal: So wie diese Krankheit von vielen beschrieben wird existiert sie definitiv nicht. Es ist eine katastrophale Beschreibung eines unbekannten Syndroms, diagnostiziert von katastrophal unwissenden Ärzten, die Ihre Patienten in katastrophale Todesangst versetzen. Es gibt weder die Diagnose für Amyotrophe Lateralsklerose, noch gibt es Menschen die Amyotrophe Lateralsklerose haben. Wenn ich hier also von Amyotrophe Lateralsklerose spreche, dann meine ich damit Menschen die nicht nur, aber unter anderem eine Belastungsstörung der motorischen Nerven haben. Viel mehr als das darf man in diese Krankheit auf gar keinen Fall hineininterpretieren.
Ursachen
Die Neurologen sind natürlich gezwungen der Sache einen Namen zu geben, das ist ja verständlich, aber bzgl. der Lebenserwartung Aussagen zu treffen ist unverantwortlich, denn die Statistik wird erst berechnet nachdem die Diagnose gestellt wurde und der Patient unter Schock sich selbst überlassen wurde. Es wird der Glaube vermittelt, dass es keine Therapie gäbe und der Patient unheilbar krank sei und somit in Angst auf den gewissen Tod warten darf. Ein "tolles" Angebot was die Schulmedizin hier anbietet. Es scheint als habe sich die Wissenschaft noch immer nicht die Frage gestellt warum manche Amyotrophe Lateralsklerose Patienten mit Ihrer Krankheit sehr alt werden. Immerhin ist den Gelehrten inzwischen aufgefallen, dass Amyotrophe Lateralsklerose keine reine Nervenkrankheit ist, sondern eine Multisystemerkrankung und nur unter Anderem eine Motorneuron-Erkrankung. Wobei die Motorneuronen innerhalb dieser komplizierten Systemerkrankung natürlich nur die Opfer und nicht die Täter sind, daher spreche ich auch nur von einer Motorneuron-Belastung und nicht von einer Motorneuron-Erkrankung. Es bringt also nichts, wenn alle wie hypnotisiert immer nur auf das Nervensystem starren. Wenn z.B. in einem Aquarium die Fische anfangen zu sterben, dann untersucht man als erstes das Wasser und nicht die Fische. Das Wasser, also die Umgebung der Nerven, wird in der Schulmedizin vollständig vernachlässigt. Die Umgebung der Nerven ist aber die Ursache und nicht der Nerv. Diese eigentlich logische Erkenntnis ist Bahnbrechend, denn nicht-neurologische Erkrankungen lassen sich garantiert besser behandeln, als eine imaginäre Nervenkrankheit. Das bedeutet also, dass mehrere Organe oder Stoffwechselsysteme involviert sind und das bedeutet auch, dass es Wahrscheinlich keinen Amyotrophe Lateralsklerose Patienten gibt bei dem genau dieselben Systeme in gleicher Weise involviert sind. Natürlich erschwert das eine Diagnose und natürlich macht das eine Therapie äußerst kompliziert, aber Heilung ist nun mal sehr kompliziert.Die Wahrscheinlichkeit, dass eine einzelne Substanz (Monotherapie) bei einer Multisystemerkrankung wie der amyotrophen Lateralsklerose eine Heilung herbeiführen wird, ist extrem unwahrscheinlich. Trotzdem werden Studien mit Monotherapien durchgeführt, welche dann aber keine ausreichend homogenen Ergebnisse erzielen. Dabei wird erwartet, dass alle Patienten gleich reagieren, was bei einer uneinheitlichen Multisystemerkrankung jedoch wenig Sinn ergibt. Auch die Pathogenese ist vielfältig, aber was genau die Patienten vereint weiß die Forschung heute nicht. Das liegt aber auch daran, dass Neurologen gar keine oder nur eine sehr schlampige Anamnese (Erfassung der Krankengeschichte) machen. Eigentlich müsste man alle potentiellen Ursachen von jedem Patienten in einer nationalen Datenbank erfassen. Dazu müssten die Patienten aber erst mal die Risiken kennen und das wäre natürlich Aufklärungsarbeit und die wird nicht von den Kassen bezahlt. Die Ursache von Amyotrophe Lateralsklerose muss man sich in etwa vorstellen wie einen bunten Wiesenstrauß; viele Blumen ergeben einen "schönen" Duft. Jeder darf sich also aus den möglichen Ursachen, bzw. Risiken, sein "Sträußchen" zusammenstellen. Einige Studien untersuchten die Ursachen etwas genauer. Zu den möglichen Risiken gehören: Gehirnerschütterung (4,32-faches ALS Risiko) [Lehman 2012], Aluminiumbelastung (Erkrankung beim Menschen durch 42-fache Bodenbelastung) [Crapper 1988], Aluminium- und Manganbelastung bei Kalziummangel (Tierversuch beim Affen) [Garruto 1989], Diabetes mellitus (Zuckerkrankheit), Typ-1 (5,4-faches Risiko) [Mariosa 2015], Autoimmunkrankheiten [Turner 2013], Chronischer Magnesium- bei gleichzeitigem Kalziummangel (Tierversuch Ratte) [Yasui 1997], Gestörte Autophagie [Lüningschrör 2016], Chronische Entzündungen (COX-2) [McGeer 2002], Schwermetalle (Quecksilber und Blei), Pestizide und andere Organophosphate und / oder organische Lösungsmittel, Elektroschock, Hirn-Trauma [Wang 2016], Mitochondriale Dysfunktion [Crugnola 2010], Quecksilberbelastung, -vergiftung [Schwarz 1996], Angststörung (3,6-faches ALS Risiko), Depression (4,4-faches ALS Risiko) [Turner 2016], Vitamin E Mangel, Mangel an mehrfach ungesättigten Fettsäuren
Selbstverständlich wurden längst nicht alle möglichen Risiken untersucht und die bisherigen Risiken gelten noch nicht als etabliert, trotzdem erkennt man an den wahrscheinlichen Risiken, dass es sich bei Amyotrophe Lateralsklerose um eine Belastungsstörung handeln muss. Muskulatur und Nervensystem leiden unter einer Gesamtlast und eine Heilung wäre somit Entlastung und Erholung. Sie wundern sich, dass ich das Wort Heilung verwende? Selbstverständlich! Nur weil es noch keine Wunderpille gibt, bedeutet das noch lange nicht, dass Amyotrophe Lateralsklerose unheilbar ist. Es beutet nur, dass die Pharmaindustrie bisher keine patentierbare Substanz gefunden hat, welche Amyotrophe Lateralsklerose behandeln kann. Zwischen Himmel und Erde gibt es jedoch mehr als gut bezahlte Neurologen die Blinde Kuh spielen.
Als etabliert gilt hingegen, dass Männer häufiger an Amyotrophe Lateralsklerose erkranken als Frauen. Die Unterschiede zwischen Mann und Frau sind natürlich zahlreich, trotzdem darf ein wenig spekuliert werden. Vergleichen wir also den Eindruck den wir aus den Risiken gewonnen haben mit dem was Männer typischerweise falsch machen: Sie haben häufiger Unfälle, sie haben häufiger Vitaminmangel, weil sie sich schlechter ernähren, sie nehmen weniger Rücksicht auf sich selbst und sie neigen eher dazu ihre eigene Belastbarkeit falsch einzuschätzen. Könnte passen, oder? Das wären definitiv bessere Voraussetzungen für eine Belastungsstörung als bei Frauen.
Amyotrophe Lateralsklerose ist weniger eine Nervenkrankheit sondern eher eine Muskelkrankheit. Der Muskel verliert an Kraft, dann tendenziell an Volumen und nur bei sehr aggressiver Oxidation oder anderen zusätzlichen biochemischen Belastungen im Muskel degenerieren möglicherweise auch motorische Nerven. Amyotrophe Lateralsklerose ist daher nur sekundär eine Nervenkrankheit. Der Muskelstoffwechsel ist quasi der "Kriegsschauplatz" für eine nur tendenziell degenerative Belastung der motorischen Nerven. Dieser Kollateralschaden kann aber vermieden werden und wenn es gelingt den Körper umfangreich zu entlasten und zu regenerieren, dann wäre theoretisch auch eine Heilung möglich.
Menschen mit Amyotropher Lateralsklerose oder anderen neurodegenerativen Krankheiten geht es nicht immer gleich gut oder schlecht. Die neurologische, symptomatische Fluktuation kann erheblich sein, sogar binnen eines Tages oder weniger Stunden. Selbstverständlich hat das nichts mit dem Verlust oder dem Zugewinn von Nervenzellen zu tun, sondern mit der variablen, der exogenen (äußeren) Belastung der Nervenzellen, bzw. mit dem variablen Schutz der Nervenzellen. [Palop 2006] Umweltgifte, Komorbiditäten, Ernährung, Lebensstil und Psyche sind hierfür die wichtigsten veränderbaren Einflüsse und müssen daher unbedingt berücksichtigt werden.
Muskelstress
Starke muskuläre Beanspruchung ist ein sehr gefährlicher neurodegenerativer Multiplikator bei Amyotrophe Lateralsklerose, weil der katabole Stoffwechsel der Muskulatur so verstärkt wird und freie Radikale vermehrt freigesetzt werden, sodass eine Art neurotoxisches Milieu im Muskel entsteht. Es wird mehr Muskelmasse zerstört und eine Regeneration bleibt aus. Eine Verbesserung der Regenerationsfähigkeit ist daher ein wichtiges Ziel bei der Therapie von Amyotropher Lateralsklerose.Benign Fasciculation Syndrome (BFS)
Faszikulationen kennt jeder als nervöse Zuckungen unter dem Auge, können aber am ganzen Körper auftreten. Sie sind zu 93% chronisch und sind meistens auf eine nicht pathogene Angststörung zurückzuführen. Häufig auftretende Symptome sind subjektive Schwäche, sensorische Symptome und Krämpfe. Alleine führt diese Symptomatik nicht zu Motorneuronerkrankungen. [Filippakis 2018] Wenn Faszikulationen also ein eigenständiges Syndrom sind, dann könnte das bedeuten, dass sie auch ein eigenständiger Teil der multisystemischen Erkrankung sind und, dass eine Angststörung eine der Ursachen von Amyotropher Lateralsklerose sein kann. Wohlbemerkt eine Ursache und keine Folge von Amyotrophe Lateralsklerose, weil Faszikulationen ein Frühsymptom bei Amyotropher Lateralsklerose sind.Eine Angststörung als eine der Ursachen von Amyotrophe Lateralsklerose? Interessant, aber stimmt das auch? Wer mit einer Diagnose von Schizophrenie, bipolarer Störung, Depression oder Angst in ein Krankenhaus eingewiesen wurde, bei dem wurde bereits im nachfolgenden Jahr häufiger die Erstdiagnose Amyotrophe Lateralsklerose gestellt (3,6-fach erhöhtes Risiko). [Turner 2016] Kommen Neurologen auf die Idee Angststörungen oder Depressionen zu Untersuchen, geschweige denn zu behandeln? Nö. Und das obwohl bis zu 54% der Amyotrophe Lateralsklerose Patienten als depressiv gelten (29% milde- und 6% schwere Fälle). [Atassi 2011]
Nur etwa 5% der Patienten mit diesem Syndrom erkranken an Amyotrophe Lateralsklerose.
Nitrosativer Stress
Neben der Zell-, DNA- und Protein-Schädigung unter der Beteiligung von Sauerstoff (Oxidativer Stress), gibt es auch den weniger prominenten sog. nitrosativen Stress, bei dem Stickstoffmonoxid beteiligt ist. Nitrosativer Stress ist besonders gefährlich, weil er zu neurodegenerativen Erkrankungen führen kann. Neurologen scheint das wenig zu interessieren, was äußerst bedauerlich ist, denn um nitrosariven Stress zu messen müsste man nur mal die M2PK-Aktivität untersuchen (Stickstoffmonoxid NO Überlastung). Der Test kostet grade mal € 25,-. Ein bekanntes Symptom von nitrosarivem Stress ist die sog. Fressnarkose, also Müdigkeit nach dem Essen. Diese Form der Müdigkeit resultiert aus einer Stoffwechselblockade welche den Transfer von Glukose in die Mitochondrien verhindert. Die Energie ist in Form von Glukose verfügbar, aber die Zellen können den Treibstoff nicht aufnehmen. Im Prinzip ist das eine Art Lähmung auf Zellebene.Aber was hat das mit Amyotrophe Lateralsklerose zu tun? 3-Nitrotyrosin dient in der Labordiagnostik als Biomarker für nitrosativen Stress und genau dieser Wert war in einer Untersuchung von Patienten mit Amyotrophe Lateralsklerose 7-fach erhöht. [Tohgi 1999]
Es sollte also niemanden verwundern, dass 3-Nitrotyrosin ganz allgemein ein Biomarker für Apoptose (programmierter Zelltod) ist. [Abu-Qare 2001]
Interessiert das Neurologen? Nö. Diese Problematik kennen nur sehr wenige Ärzte.
Cushing-Syndrom / Chronischer Stress
Cushing-Syndrom ist ein krankhaft erhöhter Cortisolspiegel (Hypercortisolämie) mit Symptomen wie:- Angstattacken
- Anhedonie (Unfähigkeit, Freude und Lust zu empfinden)
- Appetitschwankungen
- Bluthochdruck (Hypertonie)
- Erhöhter Blutzuckerspiegel
- Erhöhter Durst
- Gewichtszunahme
- Verlust der Muskelkraft (Myopathie)
- Häufiges Wasserlassen (typisch bei akuten Zuständen)
- Störung der Hypothalamus-Hypophysen-Nebennierenrinden-Achse (Hormone)
- Kognitive Beeinträchtigungen
- Motivationsschwankungen
- Kaliummangel (Muskelschwäche und Muskelkrämpfe)
- Psychosen
- Reduzierte Muskelmasse
- Rundes Mondgesicht oder aufgequollenes Gesicht
- Stammfettsucht (dünne Arme und Beine, dicker Rumpf mit „Büffelnacken“)
Cushing-Syndrom ist selten, mit einer Inzidenz von 0,7-2,4 pro Million Einwohner pro Jahr. Bei einem Bevölkerunsverhältnis von 1:1.000.000 ist das Cushing-Syndrom in etwa doppelt so selten als Amyotrophe Lateralsklerose.
Wenn ich zu einem Endokrinologen gehe und ich höre als erstes die Frage: "Was erwarten Sie von mir?", dann fallen mir eher eine Reihe lustiger Antworten ein. Muss denn der Patient wissen, was er von seinem Arzt erwarten kann? Eine gute Antwort wäre: "Ich erwarte von Ihnen, dass Sie mir sagen, was ich von Ihnen erwarten kann.", oder "Ich erwarte Heilung!". Letztlich stellte ich ihm eine Gegenfrage: "Gibt es endokrinologisch bedingte Ursachen für Muskelschwäche?". Seine Antwort kam als er auf meine Blutwerte schaute: "WOW! Ihre Sexualhormone hätte ich auch gerne!". Außer dumme Sprüche und eine fette Rechnung kann man von Endokrinologen offensichtlich nicht erwarten. Dumm nur, dass Cortisol im 24-h-Urin gemessen wird und nicht im Blut.
Bei einem Facharzt für Innere Medizin, Hämatologie und internistische Onkologie kam als Erstfrage: "Hat Ihre Frau Sie geschickt?". Diagnose war: Psychisch krank. Adenome oder Karzinome der Nebennierenrinde, welche das Cushing-Syndrom auslösen können, wurden NICHT abgeklärt. Auch ein Insulinom kann zu Muskelschwund führen. [Jaspan 1982] Seine Diagnose belegte aber, dass er Insulinome nicht kennt, sonst hätte er Muskelschwund seinem Fachgebiet zuordnen können. Rechnung für Ignoranz und Frechheit folgte. Fazit: Der Arzt kannte sich weder mit dem Fachgebiet der Neurologie aus, noch mit seinem eigenen Fachgebiet, stellte dann aber eine Diagnose aus dem Fachgebiet der Psychiartrie, obwohl das gar nicht sein Fachgebiet ist.
Ein Gastroenterologe war mal tatsächlich so schlau das morgendliche Cortisol im Blut zu messen, leider ohne die Gabe von Dexamethason am Abend davor. Diese ganze Medizin-Show heißt: Ärzte nutzen ihr Halbwissen um Geld zu verdienen.
Hypercortisolämie äußert sich anfangs durch zunehmendes Bauchfett und später durch eine mittelgradige Depression; das bedeutet, dass man noch arbeiten kann, aber man schleppt sich nur noch durch die Gegend. Man glaubt, dass die aktuellen Lebensumstände das depressive Empfinden verursachen, doch die Ursache liegt länger zurück. Das führt zu Schuldzuweisungen gegen nahestehende Personen, da Stress als Ursache unglücklicher Empfindungen nicht bekannt ist. Die aktuellen Lebensumstände haben aber mit dem Stress, den Sie schon vor Jahren zu lange toleriert haben, nichts zu tun. Durch das kontinuierliche Ignorieren Ihrer Stressbelastung verhindern Sie lediglich eine Heilung, aber Unwissenheit schützt nicht vor Krankheit. Ohne den neurologischen, Schaden würde man seine Lebensumstände viel positiver einschätzen. Die negative Lebenssituation entsteht erst nach und auf Grund von bereits erworbenen neurologischen Defekten oder vorausgehenden Traumata.
Was hat das mit Amyotrophe Lateralsklerose zu tun? Untersuchungen zeigten bei Amyotrophe Lateralsklerose Patienten einen 1,4-fach erhöhter Cortisolspiegel gemessen wurde. [Gargiulo Monachelli 2011] Interessiert das Neurologen? Nö. Man kann aber die Studie ausdrucken und einem Endokrinologen vorlegen um ihn zu belehren. Das klingt überheblich, vermeidet aber das absolut typische Gedankenmuster bei Fachärzten: "Der Patient hat ausschließlich und offensichtlich eine neurologische Krankheit. WAS WILL DER BEI MIR???". Diese Einstellung führt zu einer halbherzigen und oberflächlichen Arbeitsweise, zu mangelhafter Aufmerksamkeit und verminderter Gründlichkeit des Facharztes á la "Kasse machen und abharken". Einen derart herabwürdigenden Umgang mit Ihrer Person sollten Sie von vornherein vermeiden. Am besten fragen Sie den Endokrinologen gleich vorweg ob er das Cushing-Syndrom kennt. Wenn er ja sagt, sind Sie an der richtigen Adresse, wenn er nein sagt, fragen Sie ihn, ob er einen Spezialisten für Sie finden kann der das Syndrom kennt. Ärzte müssen den Umgang mit ihrer eigenen Unwissenheit manchmal erst erlernen. Sind sie es doch gewohnt den weißen Kittel der Allwissenheit von Göttern zu tragen.
Die circadiane Rhythmik bestimmt unsere inneren Rhythmen wie den Schlaf-Wach-Rhythmus, aber auch den Anstieg und Fall von Stresshormonen. Stresssensible Menschen reagieren auf alles mit einer zu starken hormonellen Reaktion, deshalb ist ein Psycho-Thriller am Abend, wenn die Stresshormone durch den circadianen Rhythmus herunterfahren, nicht die richtige Wahl. Wenn sie sich den Thriller trotzdem ansehen wollen, dann machen Sie eine Übung draus: Lachen Sie in bedrohlichen Scenen. Wenn Sie darüber lachen können, dann können Sie über alles lachen und das sollten Sie auch, denn Lachen ist das hormonelle Gegenteil von Angst und sagt dem Gehirn auf hormoneller Ebene das die Gefahr vorbei ist.
Paradoxerweise hat körperliche Aktivität keine negativen Folgen für die Gesundheit, obwohl der Cortisolspiegel bei körperlicher Aktivität genauso ansteigt wie bei psychischem Stress. Der wesentliche Unterschied ist, dass bei körperlicher Aktivität Dopamin ausgeschüttet wird, bei Stress hingegen nicht. [Chen 2017] Bei Amyotrophe Lateralsklerose Patienten zeigte sich eine Dysregulation der Nebennierentätigkeit mit ungewöhnlich hohem Cortisolspiegel am Abend. [Patacchioli 2004] Ein kleiner Spaziergang von 5-20 min. nach dem Abendessen könnte den Dopamin-Spiegel anheben und so möglicherweise den negativen Effekt von Cortisol maskieren. Allerdings sollten sie die körperliche Aktivität am Abend nicht übertreiben, denn Sie sollten den circadianen Rhythmus unterstützen, um von den Phasen mit niedrigen Cortisolspiegel zu Profitieren.
Andere Untersuchungen zeigten bei Amyotrophe Lateralsklerose Patienten einen Anstieg des Cortisolspiegels am Morgen (+33%). [Spataro 2015] Was auf Dauer durchaus gefährlich ist, weil der Cortisolspiegel 30 bis 45 Minuten nach dem Aufwachen schon von Natur aus am höchsten ist. Opas spießige Morgengymnastik entpuppt sich also als höchst sinnvoll, denn der morgentliche Cortisol-Schub sorgt für reichlich Glukose im Blut, welche durch etwas Bewegung genutzt werden darf. Der Cortisolspiegel hat auch ein Nachmittagstief zwischen 14 und 17 Uhr, mit einer Dauer von 15 Minuten bis 2 Stunden. [Jacobs 1996.] Diese Siesta-Pause sollte zur Regeneration genutzt werden. Z.B. nach dem Mittagessen ein kleiner Spaziergang (20-30 min) und dann 2 Stunden im Liegestuhl aktiv entspannen und meditieren. Etwa um 4:30 Uhr ist der Cortisolspiegel am niedrigsten.
Übergewichtige haben laut Untersuchungen niedrigere Cortisolspiegel als normalgewichtige Vergleichspersonen. Es ist also sinnvoll sein Gewicht zu halten während von kalorienreduzierten Diäten dringend abzuraten ist. [Travison 2007]
Stress begünstigt die zentrale Fettverteilung und macht laut Studien empfindlicher für negativen Stress und für dessen begleitende Cortisolausschüttung. Der Regelkreis zwischen Hypothalamus-Hypophyse- und Nebenniere scheint dabei schlechter zu funktionieren. [Vicennati 2000] Normaler Stress führt also zu mehr negativ empfundenen Stress. Sie kennen das: Sie haben extremen Hunger und sind schon total grantig. Das ist Cortisol! Durch aggressives Verhalten provizieren Sie aber auch negatives Verhalten Ihrer Mitmenschen. Cortisol verändert also Ihr Verhalten und auch das Ihrer Freunde und Familie. Sie erschaffen sich eine negative Realität. Ein gutes Beispiel: Ich fuhr mal mit einem Handwerker als Beifahrer mit. Plötzlich kam ein Auto von der Seite und provozierte beinahe einen Unfall. Unfassbare Dummheit im Straßenverkehr, aber der Handwerker lachte nur schallend laut und konnte kaum aufhören zu lachen. Das ist gesundes Verhalten.
Mithilfe einer funktionellen MRT wurde gezeigt, dass Personen je nach Stressbelastung unterschiedlich auf Bilder mit hochkalorischen Lebensmitteln reagieren: Bei stark gestressten Personen führten solche Bilder zu einer erhöhten Aktivität von genau den Gehirnregionen, die für Belohnung und Motivation verantwortlich sind und zu einer verminderten Funktion des präfrontalen Kortex. Chronischer Stress kann somit die Aktivität bestimmter Hirnregionen und Ernährungsverhalten hin zu schmackhaften, kalorienreichen Lebensmitteln beeinflussen.
Eine australische Studie belegt, dass der tägliche Verzehr von vier bis sieben Portionen Obst und Gemüse mit einem geringeren Auftreten von hohen Stressbelastungen in Zusammenhang steht. [Nguyen 2017]
Cortisol ist unter Stress für 73% der Gewichtszunahme verantwortlich. Primär Kohlenhydrate und gesättigte Fette werden dabei vermehrt konsumiert. [Roberts 2013]
Beruflicher Stress, z.B. durch zu hohe Erwartungen und niedrige Entscheidungskompetenz, führt zu einer erhöhten Kalorienzufuhr durch energiereiche Lebensmittel mit mehr Fett und Zucker. [Nishitani 2005] Z.B. steigt die Wahrscheinlichkeit ungesunde Snacks zu essen. [Maier 2015]
Eine sehr angenehme Art Cortisol zu sänken ist Sauna. Außerdem werden dabei auch anabole Hormone wie Testosteron und das Wachstumshormon erhöht. [Kukkonen-Harjula 1989]
Glucoseintoleranz/Insulinresistenz
Menschen mit Glucoseintoleranz haben das Problem, dass Ihre Zellen Glucose nicht mehr ausreichend aufnehmen; das Insulin kann die Zelle nur noch sehr begrenzt mit Glucose versorgen. Dieser gestörte Glukose-Stoffwechsel erhöht bei Patienten mit Typ-2-Diabetes z.B. das Alzheimer-Risiko um 65 Prozent. [Arvanitakis 2004] Das ist nicht verwunderlich, denn die Medizin kennt den Zusammenhang bereits von der diabetischen, peripheren Neuropathie, welche durch eine neurotoxische Hyperglykämie entsteht. Zu viel Glucose im Blut wirkt also wie ein Nervengift. So kam es, dass man heute bei Alzheimer auch von Typ-3-Diabetes spricht.Wenn die Glukose es nicht in die Zelle hinein schafft, dann hat die Zelle auch keine Kraft die für Sie vorgesehene Aufgabe zu erfüllen, geschweige denn sich effektiv um sich selbst zu kümmern.
Insulinresistenz ist möglicherweise sogar ein frühes Warnzeichen bereits bevor erste kognitive Symptome eintreten. [Baker 2010]
Das metabolische Syndrom ist von der bipolaren Störung und der Depression metabolisch kaum zu unterscheiden. Es handelt sich um immunentzündlichen, oxidativen und nitrosativen Stress. Genauer gesagt handelt es sich um chronische geringgradige Entzündungen, einschließlich erhöhter Mengen von pro-inflammatorischen Zytokinen und Akute-Phase-Proteinen, erhöhte Lipidperoxidation mit Bildung von Malondialdehyd und oxidierte Low-Density-Lipoprotein-Cholesterin (LDL-c), Hypernitrosylierung (starker nitrosativer Stress), verringerte Mengen an Antioxidantien, vor allem Zink und Paraoxonase (PON1), erhöhte bakterielle Translokation (Leaky Gut), erhöhter atherogener Index von Plasma und Castelli-Risikoindizes; und reduzierte Spiegel von HDL-Cholesterin (high density lipoprotein). Es handelt sich also um systemische neuro-immune, neuro-oxidative und neuro-nitrosative Stoffwechselstörungen. [de Melo LGP 2017]
Eine Studie zeigte, dass bereits Prädiabetes mit ähnlichen Risiken für periphere Neuropathie und Schweregrad der Nervenfunktionsstörung assoziiert war wie neu auftretender Diabetes. Sie fanden auch eine unabhängige Verbindung zwischen Prädiabetes und der peripheren Neuropathie und der Schwere der Nervenfunktionsstörung. [Lee 2015]
Beim Cushing-Syndrom entsteht eine Stoffwechsellage ähnlich wie bei Diabetes mellitus mit Durst und häufigem Wasserlassen. Das bedeutet, dass eine neurotoxische Hyperglykämie auch ohne Diabetes möglich ist. So entstand der Name Cushing's Neuropathy bereits 1958. [O'SULLIVAN 1958]
Unter Belastung bezieht der Muskel den Großteil seiner Energie aus dem im Muskel lokal gespeicherten Glykogen. Glukose aus der Nahrung kann einen geringen Beitrag leisten und auch die Leber steuert mit andauernder Belastung zuhnehmend mehr Glukose bei. Entscheidend ist jedoch das Glykogen im Muskel. Ist der Speicher aufgebraucht lässt die Kraft nach. [Bergström 1967] Kraftausdauer und Ausdauer sind also also hochgradig davon abhängig ob und wieviel Glykogen im Muskel gespeichert wurde.
Die Glykogen-Synthese ist von außerordentlicher Bedeutung. Forscher schalteten bei Mäusen das Gen aus welches für die Glykogen-Synthese verantwortlich ist. Das Ergebnis war beeindruckend: Hohe Blutzuckerspiegel, hohe Insulinspiegel, Insulinresistenz und Glucose-Intoleranz. [Andrikopoulos 2016] Der Grund von schneller Ermüdung liegt also in einer niedrigen Glykogen-Synthese.
Für die Glykogen-Synthese brauchen wir Insulin, glukosetolerante, insulinsensible Zellen und körperliche Aktivität. [Yeaman 2001] Wir drehen uns also im Kreis, denn letztlich ist irgendwie alles abhängig von körperlicher Aktivität. Wir müssen uns also bewegen um Insulinresistenz zu überwinden und um mehr Glykogen zu speichern. Die Frage ist nur wie trainieren wir ohne zu großen Schaden anzurichten und wie bauen wir in der Regenerationsphase unsere Muskulatur maximal auf.
Hyperglykämie
Hyperglykämie kann zu partiellen Lähmungserscheinungen führen. Das bedeutet nicht, dass man eine Cola trinkt und dann z.B. die Arme nicht mehr heben kann, aber immerhin eine Reduktion der Kraft und der Kraftausdauer. Bei bereits sehr geschwächten Menschen kann es sich jedoch anfühlen wie eine vollständige Lähmung, was wiederum Panikattacken auslösen kann. Die Lähmung geht nach einigen Stunden wieder zurück, aber es Zeigt wie die Ernährung das Nervensystem schädigen kann. Es handelt sich um langsame progressive strukturelle Anomalien mit Entzündungen und Neurodegeneration. Die Ursache ist oxidativer Stress, aber wie genau die Schädigung molekular abläuft ist noch unklar. [Kumar 2017]Ein wesentlicher Unterschied zwischen Nerven- und Muskelzellen ist, dass Muskelzellen Glukose über Insulinrezeptoren aufnehmen und Nervenzellen der Hyperglykämie eher ausgeliefert sind - ein Phänomen, das als Glukose-Neurotoxizität bezeichnet wird. [Kumar 2017]
Raffinierter Zucker wird schnell in den Blutkreislauf aufgenommen; ein Phänomen, auf das der menschliche Körper evolutionär offenbar nicht vorbereitet wurde. Ein abrupter Anstieg der Blutzuckerwerte kann dazu führen, dass die Bauchspeicheldrüse eine übermäßige Menge an Insulin freisetzt, was zu einer reaktiven Hypoglykämie und einem kompensatorischen Anstieg von Adrenalin und Cortisol führen kann. [Yudkin 1978] Wer trotzdem auf Zucker nicht verzichten will, dem empfehle ich Melassesirup, denn Melasse enthält viel Chrom und Chrom soll die Insulinrezeptoren sensibilisieren und den Transfer von Glucose in die Zellen erleichtern. Chrommangel kann zu Nervenstörungen (Neuropathie) führen.
Nicht nur diabetische Blutzuckerwerte, sondern auch hoch-normale Werte könnten sich ungünstig auf das Hirnvolumen von älteren Menschen auswirken. [Walsh 2017] Wenn Nervenzellen ohnehin schon multisystemisch belastet sind, dann reicht möglicherweise schon ein hoch-normaler Blutzuckerspiegel um Schaden anzurichten.
Eine wirklich tolle Studie untersuchte antioxidative Gewürze (schwarzer Pfeffer, Zimt, Nelken, Knoblauch, Ingwer, Oregano, Paprika, Rosmarin und Kurkuma) im Vergleich zu Placebo, bei fettreicher Nahrung (1000 kcal, 45 g fat). Gewürze reduzierten nach der Mahlzeit die Triglyceride um 31%, wenn nach der Mahlzeit eine Ruhepause folgte, unter Stress hatten die Gewürze diesen Effekt jedoch nicht. Auf Glukose oder Insulin hatten die Gewürze keine Wirkung. Akuter Stress hingegen erhöhte Glucose oder Insulin jedoch beide signifikant (mittlerer Anstieg um 47% bzw. 19%). [McCrea 2014] Das ist eine beeindruckende Darstellung wie psychischer Stress krank machen kann. 47% mehr Glukose durch Stress. So entsteht das metabolische Syndrom und später Diabetes oder Herz-Kreislauf-Erkrankungen. Ärzte scheint das aber nicht sonderlich zu interessieren. Daran erkennt man auch wie massiv die Stressreaktion unseres Körpers ist. Die Studie zeigte auch einmal mehr wie stark nach dem Essen, unter Stress der Blutruck ansteigt.
Wer zum Frühstück Weißbrot isst, der kann bereits in eine Hyperglykämie rutschen. Daher empfehle ich auf Bio-Vollkorn umzusteigen (z.B. glutenfreies Chia- oder Quinoabrot). Außerdem empfiehlt es sich morgens weniger Brot und dafür ein Ei und Obst zu essen. Dadurch steigt der Insulinspiegel nicht so hoch an und die Kohlenhydrate werden langsamer verdaut, sodass eine Hyperglykämie vermieden wird. Das gleiche gilt übrigens auch für Kartoffeln die nur als Beilage gegessen werden sollten. Viele können morgens Eiweiß besser verdauen, daher ist das Frühstücksei eine gute Strategie um die Proteinzufuhr zu erhöhen. Eine optimale und Magen-Darm schonende Eiweißverdauung ist von äußerster Wichtigkeit. Es geht nicht darum einfach nur mehr Eiweiß zu essen, es geht darum Eiweiß vollständig zu verdauen, damit er auch vollständig vom Körper aufgenommen werden kann. Mann könnte auch sagen, es geht darum das Stuhlvolumen zu verringern. Das Ei sollte nach den Kohlenhydraten gegessen werden, weil sich dann bereits Magensäure gebildet hat. Magensäure bildet sozusagen eine "Willkommenskultur" für das Eiweiß. In Italien isst man nicht ohne Grund die Kohlenhydrate als Pasta zur ersten Hauptspeise und das Fleisch erst danach, als zweite Hauptspeise. Ein Ei auf nüchternen Magen zu essen löst bei manchen Menschen Sodbrennen oder brennenden Hustenreiz aus. Solche Symptome der Proteinverdauung sollten sehr ernst genommen werden, denn ein Proteinmangel fällt natürlich nicht vom Himmel und muss unbedingt vermieden werden. Eine effiziente Proteinverdauung ist die primäre Grundlage für eine maximale Resorption von lebenswichtigen, essentiellen Aminosäuren im Darm. Ein Frühstücksei gehört daher zum täglichen Pflichtprogramm.
Manche werden wegen dem Cholesterin im Eigelb Bedenken haben, doch diese Befürchtungen sind unbegründet, denn hohes Cholesterin erwies sich in Studien eher als Vorteilhaft und Statine sogar als nachteilhaft. Cholesterin wird z.B. benötigt um anabole Hormone zu synthetisieren.
Hypermetabolismus
Hypermetabolismus bedeutet dass der Energieverbrauch bei Amyotrophe Lateralsklerose Patienten um durchschnittlich 10% höher ist als bei gesunden Vergleichspersonen. [Desport 2001] Das ist ein bischen so als ob Sie irgendwann losgelaufen sind und dann, obwohl Sie längst aufgehört haben zu laufen, der Körper nicht mehr bemerkt, dass Sie bereits ruhen und immer weiter so tut als würden Sie für den Rest Ihres Lebens immer weiterlaufen. Auch der Cortisolspiegel steigt dadurch öfter an Sie schneller unterzuckern.Patienten mit amyotropher Lateralsklerose leiden also an Hypermetabolismus. Die Frage ist dabei nur, wer war zuerst da, die Henne oder das Ei? In meinen Augen ist der Hypermetabolismus sowohl an der Entstehung, als auch am Krankheitsverlauf von Amyotrophe Lateralsklerose beteiligt und nicht umgekehrt. Das erscheint mehr als logisch sobald man sich die Ursachen von Hypermetabolismus anschaut: Durch Gewebeschäden, wie bei einem chirurgischen Eingriff oder Brandverletzungen, Traumata, Schock, Infektionen oder anderen entzündlichen Zuständen, scheint es einen anhaltenden Hypermetabolismus zu geben, der eine abnormale metabolische Regulation verursacht, zu L-Arginin-Mangel führt, die Entzündungsreaktion verlängert und von Immunsuperession begleitet wird. Im Extremfall kann ein solcher Zustand zu multiplem Organversagen führen. [Cerra 1991] Na, wenn das kein De ja-vue Erlebnis ist; gute Freunde trifft man immer wieder. Bei Hypermetabolismus muss man also sehr ausführlich auf Entzündungen untersuchen und bei Auffälligkeiten nach der Ursache suchen. Motorneuronen sind garantiert nicht die Ursache.
Während eines Traumas entsteht Stress, Hypermetabolismus, Störung der Proteinsynthese und Katabolismus. Anhaltender mäßiger bis starker Stress verursacht Nährstoffmängel, Mangel an essentiellen viszeralen Proteinen (Unterernährung), Beeinträchtigung der Immunfunktion, Schwächung der Entzündungsreaktion und Störung der Wundheilung. Die erhöhte Zufuhr von L-Arginin ist vorteilhaft für die Immunfunktion, da die Aminosäure für Wachstum notwendig ist, außerdem wurde gezeigt, dass durch L-Arginin T-Helferzellen vermehrt werden, die Lymphozytenreaktion auf Mitogene erhöht wird, während T-Suppressorzellen verringert werden. [Van Way 1991]
Hypermetabolismus bewirkt nicht nur einen höheren Verbrauch von Glukose sondern auch einen höheren Verbrauch von Glutamin durch Glukoneogenese. Dadurch steht weniger Glutamin für die Proteinsynthese und den Einbau in Makromoleküle zur Verfügung.
Der erste wichtige Schritt bei der Synthese von Glukose (Blutzucker) hängt von einem Enzym ab, das Biotin enthält. Die Synthese von Glukose ist notwendig, um den Blutzuckerspiegel konstant zu halten und Hypoglykämie zu verhindern.
Hypoglykämie (Unterzucker)
Die typische Hypoglykämie entsteht jedes Mal dann, wenn wir über längere Zeit nicht essen. Abhängig davon was und wieviel wir essen, kann das sehr schnell passieren oder erst nach vielen Stunden. Sinkt der Insulinspiegel wegen dem niedrigen Blutzucker dann zu stark ab (<80mg/dl), schaltet sich der metabolische Katabolismus ein, indem die Bauchspeicheldrüse von Insulin- auf Glukagon-Produktion umschaltet. Daraufhin wird Adrenalin und Cortisol von den Nebennieren abgegeben. Glukagon gilt als das wichtigste katabole Hormon des Körpers. Insulin senkt den Blutzucker durch Speichern von Glukose (anabol) in den Zellen, während Glukagon den Blutzucker erhöht, indem es das in der Leber gespeicherte Glykogen in Glukose umgewandelt und dann in den Blutkreislauf freigibt (katabol). Die Leber liefert bei gesunden Menschen für ca. 24 Std. Glukose ("Notstrom"), doch sobald Sie körperlich aktiv werden, muss die Leber das 7-fache an Glukose bereitstellen. Die Gluconeogenese fällt wesentlich schneller ab auf ein kritisches Niveau. [Iwashita 2005]Ein Beispiel: Sie schlafen zu lange (ca. 10 Stunden) und fühlen sich beim Aufwachen gestresst und körperlich erschöpft als wären Sie gar nicht zu Bett gegangen. Was Sie nicht wissen, ist, dass im Schlaf Ihr Blutzuckerspiegel zu stark abgesunken ist und Sie daher von einem starken Cortisol-Schub geweckt wurden. Hypoglykämie ist eigentlich etwas Gesundes, wir kennen die Vorteile vom Fasten, aber für Nervenkranke ist metabolischer Stress ein Problem, denn Überstimulierung von Nerven ist genau der Zustand der zum Krankheitsbild eher beiträgt. Die Hälfte aller hypoglykämischen Ereignisse treten nachts auf. [The DCCT Research Group 1991], [De Feo 1986] 7-8 Stunden Schlaf muss daher ausreichen und ein spätes Abendessen (2-3 Stunden vor der Nachtruhe) ist sehr anzuraten. Körperliche Aktivität auf leeren Magen ist absolutes Tabu. Im Gegenteil, man sollte vor und nach dem Spazieren gehen Kohlenhydrate und Proteine essen, um erst den Katabolismus zu minimieren und nach dem Sport den Anabolismus zu maximieren. 30 bis 50 g Kohlenhydrate mit 5-10 g Protein vor dem Training kann den belastungsinduzierten Katabolismus verringern, während 80 bis 120 g Kohlenhydrate mit 15 bis 20 g Protein innerhalb von 2 Stunden nach dem Training ein anaboleres Hormonprofil und Glykogen-Synthese erzeugen. [Kreider 1999]
Nur weil man etwas gegessen hat bedeutet das noch lange nicht, dass man leistungsfähig ist, denn die Glukose muss es erst durch die Zellmembran in die Muskelzelle schaffen und das ist absolut nicht selbstverständlich. Bei Insulinresistenz z.B. entsteht eine intrazelluläre Hypoglykämie trotz hohen Blutzuckerspiegels. Chronisch hohe Cortisolspiegel führen z.B. zu Insulinresistenz. [Rizza 1982]
Patienten mit amyotropher Lateralsklerose verbrauchen im Ruhezustand durchschnittlich 10% mehr Energie als gesunde Vergleichspersonen. [Desport 2001] Das bedeutet natürlich, dass man bei so einem Hypermetabolismus viel schneller in den Unterzucker hineinrutscht. Und genau das scheint für Amyotrophe Lateralsklerose Patienten äußerst gefährlich zu sein: Unterernährte Amyotrophe Lateralsklerose Patienten haben ein 7,7-fach erhöhtes Sterberisiko [Desport 1999] und ein um 30% erhöhtes Sterberisiko pro 5% Gewichtsreduktion. [Marin 2011] D.h. dass der Muskel ohne ausreichend Energiezufuhr schneller abbaut. Es wird quasi alles in den Ofen geworfen - Fett, Glukose, Muskelproteine - Hauptsache die Maschine läuft weiter auf Hochtouren. Regelmäßig und viele kleine Portionen essen, nicht zu lange schlafen und sich NIEMALS überanstrengen sing daher von kritischer Bedeutung.
Anders als die Muskelzelle kann eine Nervenzelle Ihre Energie nicht aus Fettsäuren gewinnen. Im ruhenden Muskel sind Fettsäuren sogar der Hauptbrennstoff und erfüllen 85% des Energiebedarfs. Unsere Muskulatur hat einen eigenen Glykogen-Vorrat von ca. 1200 kcal bzw. 5000 kJ (ca. 500 g, Leber: 100 g). Tatsächlich sind etwa drei Viertel des gesamten Glykogens des Körpers im Muskel gespeichert. Dieses Glykogen wird leicht in Glukose zur Verwendung in Muskelzellen umgewandelt. Dem Muskel fehlt wie dem Gehirn das Enzym Glukose-6-Phosphatase, und daher wird Glukose nicht exportiert. Vielmehr reserviert der Muskel seine Glukose als bevorzugten Treibstoff für physische Belastungsphasen. Das ist sehr wichtig hervorzuheben, denn damit wird deutlich, dass die Glykogenspeicher der Muskulatur für den katabolischen Stoffwechsel nicht zur Verfügung steht. Beim Katabolismus geht es also sofort ans Eingemachte; dem Muskelproteinen. An irgend einer Stelle während der Evolution war diese reservierte Muskelglukose von entscheidendem Vorteil, denn wer diese Energiereserve hatte, der konnte zu jedem beliebigen Zeitpunkt weglaufen und verhinderte so gefressen zu werden.
Der hypoglykämische Katabolismus ist unnötig und sollte daher vermieden werden. Wer bereits erhöhte Stresshormone hat und dann zusätzlich noch unterzuckert, der hat eine unnötig hohe Cortisol-Belastung, außerdem agieren Cortisol und Glukagon über verschiedene Mechanismen. Das bedeutet nicht, dass die Leber mehr Glukose produziert, aber Cortisol ist gefährlich, weil es die Gluconeogenese aktiviert, bei der Glukose aus Muskelproteinen und anderen Nicht-Kohlenhydraten gebildet wird. Wer bereits chronisch gestresst ist bekommt jetzt ein Problem, denn er hat seine muskulären Proteinreserven bereits aufgebraucht und konnte nicht ausreichend regenerieren. Wenn dann der Cortisolspiegel weiter durch Hypoglykämie und/oder körperliche Aktivität anstiegt, startet Cortisol einen Angriff auf den Muskel - das Auto fährt dann quasi nur noch auf den Felgen - Muskelatrophie ist die Folge. Dabei wird der Muskel zur Energiegewinnung abgebaut: Glutamin wird aus der Muskulatur abgerufen und in Glukose umgewandelt. Danach werden auch andere Aminosäuren aus der Muskulatur abgerufen und in Glutamin umgewandelt und dann ebenso in Glukose umgewandelt. Bei Glutamin-Mangel wird also der Muskel sofort substantiell angegriffen.
In mikroskopischen Untersuchungen bei Mäusen stellten Forscher fest, dass Glukosemangel zu einer Störung der synaptischen Funktion führte und zwar auf Grund einer Zellschädigung durch ein Protein. [Lauretti 2016]
Patienten die Koffein erhielten zeigten Hypoglykämie-Symptome (Zittern, Schwitzen, Herzklopfen und Angstzustände), wenn der Plasma-Glukosespiegel von 90 mg / dl auf 68,5 mg / dl gesenkt wurde. Bei der niedrigeren Glukosekonzentration waren die Serumkonzentrationen von Adrenalin, Cortisol und Wachstumshormon bei Koffein signifikant höher als bei den Patienten die nur Placebo erhielten, was darauf hindeutet, dass Koffein die biochemische Reaktion auf milde Hypoglykämie erhöht. [Kerr 1993] Insbesondere bei chronischem Stress, plus Hunger, plus Koffein steigen Adrenalin- und Cortisolspiegel besonders hoch an.
Permeabilität des Darms
Viele Studien belegen, dass Nervengifte bei Amyotrophe Lateralsklerose eine Rolle spielen. Überwiegend handelt es sich um Gifte die aus der Umwelt und der Landwirtschaft stammen wie z.B. Methylquecksilber oder neurotoxische Pestizide. Die über die Nahrung zugeführten Gifte reichen i.d.R. jedoch nicht aus um eine degenerative Krankheit zu verursachen. Es muss also neben Quecksilber, PCB und Pestiziden noch andere konkrete Neurotoxine oder pathogene Zustände geben die neurotoxisch wirken.Der gesunde Darm hat eine starke Barrierefunktion und sorgt dafür, dass nur die kleinsten, ausreichend reduzierten Moleküle in den Blutkreislauf gelangen. Der Darm kann jedoch, ähnlich wie bei einem Sonnenbrand oder einer Hautverbrennung, beschädigt werden und so für größere, toxische Makromoleküle, wie neuroaktive Peptide und neurotoxische Endotoxine (äußere Membran gramnegativer Bakterien), durchlässig werden. Das Resultat ist nicht weniger als eine chronische Vergiftung und Entzündung des Körpers; eine krankhafte intestinale Hyperpermeabilität des Darms oder (auch Leaky-Gut-Syndrome genannt). Mehr als 12.000 Studien wurden hierzu bereits durchgeführt und publiziert, leider ohne dass die Schulmedizin das Thema aufgegriffen hat.
Verblüffend ist, dass es längst zahlreiche Ursachen für eine solche erhöhte Darm-Permeabilität gibt die in der Medizin auch sehr gut beschrieben wurden. Es ist daher nicht verwunderlich, dass Leaky-Gut eine pathophysiologische Rolle bei neurodegenerativen Krankheiten wie Alzheimer zugeschrieben wird. [Köhler 2016], [Hu 2016]
Tight Junctions
Bei gesunden Menschen verbinden sog. Tight Junctions die Zellen der Darmoberfläche miteinander und bilden einen sehr engen Tunnel zwischen Darm und Blutkreislauf. Weil wasserlösliche Stoffe die lipophile Zellmembran der Darmzellen nicht passieren können, ist diese parazelluläre Route über Tight Junctions ein wichtiger Weg für den Transport von Wasser und wasserlöslichen Stoffen wie Mineralien. Diese Tight Junctions sind bei gesunden Menschen 1-2 Mikrometer groß. Das ist groß genug für kleine Moleküle und klein genug um die meisten Bakterien abzuwehren (zum Vergleich: rote Blutkörperchen sind 8 Mikrometer groß). Bei Leaky-Gut sind diese Zwischenräume jedoch 200-300 Mikrometer groß. Konkret zwingen wir damit das Immunsystem unseres Körpers unser Essen zu verdauen und große Mengen von Bakterien abzuwehren. Das ist natürlich ein völlig unberechenbarer Zustand. Bakterien, Antigene und Toxine und unverdaute Makromoleküle führen zu zahlreichen potentiell gefährlichen Immunreaktionen des Körpers. [McGough 2016]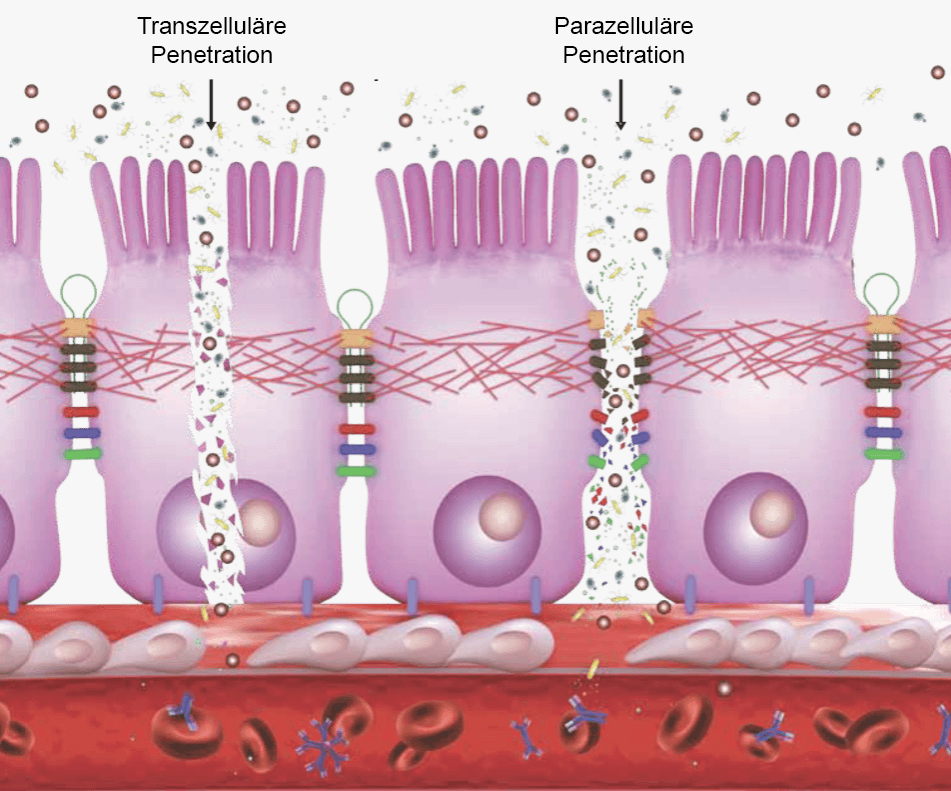
Solche systemischen Entzündungsprozesse können wiederum zellschädigend wirken. Man geht davon aus, dass die Aktivierung von Entzündungsprozessen innerhalb des Gehirns zu einer verminderten neurotrophischen (auf die Nerven einwirkend) Unterstützung und zu einer veränderten Glutamatfreisetzung / Wiederaufnahme sowie zu erhöhtem oxidativen Stress beiträgt, was zu Exzitotoxizität und Verlust von Glia-Elementen führt. [Miller 2008] Bei 86 Milliarden Glia-Zellen könnte man glauben, dass man 2 Leben braucht um ein ernsthaftes Problem zu bekommen, doch genau dies Zellen sind das Problem bei 2 der etablierten Ursachen von Amyotrophe Lateralsklerose: Astrozyten (gehören zur Gruppe der Gliazellen) tragen zur Pathologie der sporadischen amyotrophen Lateralsklerose bei. Die Dysfunktion glialer Glutamattransporter verursacht einen Anstieg der extrazellulären Glutamatkonzentrationen und exzitotoxischen neuronalen Schaden. Bei erblichen Formen von Amyotrophe Lateralsklerose führen Mutationen der Cu / Zn-Superoxid-Dismutase (SOD1) zu oxidativem Stress und anormaler Biochemie in Motoneuronen und Astrozyten. [Seifert 2006] Chronische Entzündungen sind also ein äußerst ernstzunehmendes Problem. Interessiert das Neurologen? Nö. Interessiert das andere Ärzte? Nö.
Chronischer Stress & Leaky gut
Psychologischer und körperlicher Stress kann zu einer Vielzahl von Veränderungen in der normalen gastrointestinalen Funktion führen, einschließlich Veränderungen in der Darmmotilität und Permeabilität, sowie Veränderungen in der Ionen-, Flüssigkeits- und Schleimsekretion und -absorption. [Groschwitz 2009]Die hormonelle Stressreaktion des Körpers führt zur Vergrößerung der Tight Junctions [Vanuytsel 2013], [O'Mahony 2010] um dem Körper mehr Wasser und Natrium zur Verfügung zu stellen. [Levens 1981] Evolutionär ist dieser Zustand jedoch ausschließlich für äußerst seltene überlebenskritische Situationen vorgesehen wie Kampf oder Flucht. Das war bei Primaten ein enormer Vorteil, denn die Lebewesen die mit dieser Genetik nicht ausgestattet waren dehydrierten auf der Flucht schneller, ermüdeten somit vorzeitig und wurden dann eher aufgefressen. Wenn man aufgefressen wird, dann ist es natürlich egal ob Pathogene über die Tight Junctions einmarschiert sind, der Sebelzahntieger ließ es sich garantiert schmecken. Wer aber überlebte, der hatte sehr viel Zeit sich von der systemischen Stressbelastung zu regenerieren. Das zeigen Untersuchungen an Affenfamilien in Afrika.
Das Allgemeinwissen über mögliche gesundheitliche Konsequenzen von chronischem Stress ist heute quasi inexistent. Vielmehr ist man heute davon überzeugt, das Vitaminpillen und Wellness den Körper regenerieren. Selbst Ärzte kennen die genauen Zusammenhänge nur sehr selten. Tatsächlich ist aber jede chronische Stresssituation ein Problem vor dem wir besser weglaufen sollten, bzw. welches wir schnell bekämpfen sollten. Wer z.B. Angst um seinen Job hat der sollte sich schleunigst einen neuen suchen oder in seiner Gewerkschaft aktiv tätig werden. Das sind energetische Laufrichtungen die zu einer solchen Stresssituation passen. Wer im Job Dauerstress hat, der muss sich täglich nach der Arbeit regenerieren (oder besser morgens, mittags und abends). Wer dann jedoch auch noch mit dem Lebenspartner Probleme hat, wie z.B. bei Trennungsängsten, der regeneriert nicht mehr. Das hat direkten Einfluss auf die Neuroneogenese im Hippocampus und kann so Depressionen verstärken. [Lucassen 2008]
Alkohol
Stellen Sie sich vor Sie trinken ein Glas Wein und anders als sonst fühlen Sie sich danach als hätten Sie eine ganze Flasche getrunken und haben das Gefühl als fangen Sie bereits an zu lallen. STOP! Sie haben eine schwere systemische Belastung.Alkohol erhöht die Lipidperoxidation in der Leber und verursacht drastische Veränderungen in den antioxidativen Abwehrsystemen. Insbesondere werden die Aktivitäten von hepatischer Superoxiddismutase (SOD), Glutathionperoxidase (GPx) und Katalase (CAT) durch Alkohol reduziert. [Kasdallah-Grissa 2006] Die spontane Verschlechterung der neurologischen Symptomatik gibt uns eindeutige Hinweise auf den Krankheitszustand. Superoxiddismutase reagiert z.B. mit Stickstoffmonoxid (NO) und währt so nitrosativen Stress ab. Der Alkohol reduziert die Entgiftungskapazität, sodass der nitrosativen Stress nicht mehr abgewehrt werden kann, die neurologische Symptomatik wird verstärkt. Dieser Zusammenhang zeigt auch noch mal wie wichtig die Gesundheit der Leber bei Amyotrophe Lateralsklerose ist.
Alkohol erhöht den bakteriellen Wachstum im Darm und die Permeabilität des Darms. [Leclercq 2014], [Bala 2014]
Akute Alkoholzufuhr kann die Permeabilität sogar potenzieren, denn im Darm entsteht aus Alkohol ein Gift namens Acetaldehyd [Seitz 1998] und Acetaldehyd kann wiederum die Darmpermeabilität erhöhen. [Atkinson 2001]
Das Resultat ist ein Anstieg von Endotoxinen im Plasma. [Mathurin 2000]
Außerdem sinkt die Kapazität der Leber Endotoxine zu neutralisieren, weil sie zusätzlich den Alkohol entgiften muss. [Fukui 1995] Endotoxine führen so zu Entzündungen.
Bei Ratten mit Brandverletzungen (Glutamin-Mangel) entstand bei gleichzeitiger Alkoholintoxikation ein noch fataleres Szenario: Erhöhte Permeabilität, Abnahme der intestinalen T-Lymphozyten und Zunahme der Bakterien in den Lymphknoten der Bauchhöhle. [Purohit 2007]
Die stärksten Hangover-Reaktionen zeigten sich übrigens besonders nach dem Genuss von Bourbon Whiskey. Am verträglichsten war dagegen Wodka. Allerdings war dieser Zusammenhang eher dem Stoffwechsel von Methanol zu Formaldehyd und Ameisensäure zuzurechnen. [Chapman 1970]
Candida
Candida wird in der Fachwelt nur selten ernst genommen, möglicherweise weil eine geringe Besiedelung des Darms bei 70% der gesunden Menschen als normal gilt. Die Realität ist, dass Candida im Stuhl gar nicht untersucht wird. Diese Ignoranz der Ärzte ist natürlich wieder eine dieser verpassten Chancen, denn die Ursache einer Candidiasis kann wohl kaum ein Motorneuron sein und das Abklären der Ursache könnte nützliche Informationen liefern. Dabei kennen die Neurologen die Infektionsanfälligkeit von Patienten mit Amyotrophe Lateralsklerose sehr gut, denn es ist kaum zu übersehen, dass die typische Todesursache NICHT Muskelschwund ist sondern eine Lungenentzündung. Aber selbst das reicht den Forschern nicht aus um eine Immunschwäche als mögliche Ursache zu vermuten.
Bei Asthmapatienten, die regelmäßig Cortison inhalieren, ist eine Candidiasis wohlbekannt. [Aun 2009] Hier schließt sich somit der Kreis: Stress ⇒Hypercortisolämie⇒Immunschwäche⇒Candidiasis.
Man möge meinen, dass man den Neurologen nicht vorwerfen kann Pilzinfektionen fahrlässig zu ignorieren, denn ein Pilz kann ja nichts mit dem Nervensystem zu tun haben. Weit gefehlt! Bereits seit 1970 ist die zerebrale Candidiasis bekannt; also eine Pilzinfektion im Gehirn. [Black 1970] Die Candidiasis wiederum gilt als Ursache für weitere Krankheiten.
Natürlich ist eine Candida Infektion keine eigene Krankheit, sondern eher das Resultat einer Immunschwäche. Trotzdem ist ein solcher Pilzbefall weit mehr als nur ein Symptom. Eine starke Besiedelung des Darms hat auf Dauer zahlreiche, gravierende gesundheitliche Folgen. Eine der problematischen Folgen der intestinale Candidiasis sind Lebensmittelallergien. In Tierversuchen wurde das unter anderem mit der Hyperpermeabilität des Darms begründet [Yamaguchi 2006].
In seiner Pilzform produziert Candida Abfallprodukte, die sogenannten Mykotoxine wie z.B. Acetaldehyd. Acetaldehyd ist als Abbauprodukt von Ethanol (Trinkalkohol) für dessen toxische Wirkung verantwortlich. Acetaldehyd kann die Darmpermeabilität erhöhen, indem die Tyrosinphosphorylierung von engen Verbindungsstellen und Adhärenten-Junctionproteinen erhöht wird. [Atkinson 2009] Menschen mit einer Candidiasis vertragen Alkohol daher wesentlich schlechter, weil die Leber bereits mit Acetaldehyd chronisch vorbelastet ist.
Acetaldehyd hemmt die Funktion der Lymphozyten, sodass Candida sein eigenes überleben fördert. [Roselle 1982]
Allergische Reaktionen können getriggert werden [Schulze 2009], chronische, genitale Pilzinfektionen können die Folge sein [Lin 2011], Geschwürige Dickdarmentzündung durch Abgabe von toxischem Schwefelwasserstoff (H2S) [Schulze 2009].
In Gegenwart von Acetaldehyd wird Dopamin in Salsolinol umgewandelt. Salsolinol ist ein Neurotoxin, das an der Apoptose von dopaminergen Neuronen beteiligt ist. Daher gilt eine chronische, polysystemische Candidasis als mögliche Ursache für Morbus Parkinson. Epp 2006] Eine polysystemische Candidasis ist z.B. bei genitalen Pilzinfektionen über den Harnweg erkennbar.
Wenig verwunderlich ist, dass chronischer Stress bei Frauen eine Candida-Vaginitis begünstigt. [Ehrström 2004]
Bei Alkoholkranken werden Mangelerscheinungen von Zink, Retinol, Magnesium, Vitamin B1, B6 und Glutathion direkt auf Acetaldehyd zurückgeführt. Jurnak 2015]
Ähnliche Mangelerscheinungen wurden bei Patienten mit Candidasis beobachtet. Galland 1983]
Candida verursacht außerdem Entzündungen im Darm was wiederum die Ausbreitung von Candida begünstigt. [Kumamoto 2011], [Jawhara 2008]
Schwefelwasserstoff
Ein weiteres Nervengift das von Candida abgegeben wird ist Schwefelwasserstoff (H2S). Die Toxizität von H2S ist vergleichbar mit der von Cyanwasserstoff oder Kohlenmonoxid. Es bildet eine komplexe Verbindung mit Eisen in den mitochondrialen Cytochromenzymen und verhindert so die Zellatmung. [Ramasamy 2006] Die H2S Langzeit-Einwirkung unter niedrigen Dosen kann zu Müdigkeit, Appetitlosigkeit, Kopfschmerzen, Gereiztheit, Gedächtnisschwäche und Konzentrationsschwäche führen.Traumatischer Schock
Ein traumatischer Schock, z.B. durch einen Auto- oder Motorradunfall, erhöht die Darm-Permeabilität. [Kompan 2001], [Hollander 2000] Wer Opfer eines schweren Unfalls wurde, der wird, sobald er wieder aufrecht stehen kann, aus dem Krankenhaus schleunigst entlassen. Der Patient ist auf sich allein gestellt und wird i.d.R. nicht über mögliche Spätfolgen oder unmittelbare psychische Folgen des Traumas, aufgeklärt. Da kann man zusammengekauert in einer Ecke herum heulen, bestenfalls bekommen Sie einen zweiten CT und dann ab dafür! Durch die unbehandelte Darm-Permeabilität entstehen dann Nahrungsmittelunverträglichkeiten. Fruktoseintoleranz ist ein typisches, initiales Beispiel. Eine posttraumatische Belastungsstörung kann vom Patienten selbst nicht erkannt werden und bleibt so über viele Jahre bestehen. Es ist hinreichend bekannt, dass ein so verursachter chronischer Stress, sowohl die Darmbarrierefunktion, als auch die Darmmikrobiota schädigt. [Leclercq 2016]Eine Gehirnerschütterung gilt als Risikofaktor für Amyotrophe Lateralsklerose (ALS). [Wang 2016] Wahrscheinlich kommt es aber darauf an wie, oder ob überhaupt eine Gehirnerschütterung behandelt wurde. Nehmen wir mal an Sie sind Opfer eines Auffahrunfalles und Ihr Kopf schlägt mit Wucht auf die Nackenstütze. Sie rufen die Polizei und verlangen einen Alkoholtest, doch die Polizei sagt zu Ihnen, ob das denn sein müsse. Der Täter wird umsorgt und Ihr Gesundheitszustand ist für die Polizei irrelevant. Somit wird kein Krankenwagen gerufen und Ihre Gehirnerschütterung bleibt unbehandelt und initiiert so eine lange Periode nitrosativen Stresses. Interessiert das die Neurologen bei der Anamnese? Nö.
Glutensensibilität
Nehmen wir mal an, dass Sie wegen einer Wirtschaftskrise mittags nicht mehr im Restaurant essen, sondern sich Weizenbrot und Belag aus dem Supermarkt um die Ecke holen. Nach dem Essen fallen Sie in das Nachmittagstief und werden müde. STOP! Sie haben eine Glutensensibilität! Das kann ich guten Gewissens einfach mal behaupten. Denn wenn Sie zum Arzt gehen, dann ist es sehr unwahrscheinlich, dass dieser eine Zöliakie überhaupt erkennt (und eine Glutensensibilität schon gar nicht). Die typischen Arztdiagnosen für Zöliakie sind Reizdarmsyndrom (37%), psychische Störungen (29%) und Fibromyalgie (9%). Müdigkeit nach dem Essen ist vollkommen anormal und muss sofort untersucht werden. Die typischen Symptome von Zöliakie sind Müdigkeit (82%), Bauchschmerzen (77%), Blähungen (73%) und Anämie (63%). [Zipser 2003]Unentdeckt entwickelt man später dann sogar Angstzustände und Depressionen. Leider denkt niemand bei psychischen Symptomen an eine physiologische Ursache. Der Psychotherapeut sieht das selbstverständlich genauso und therapiert fröhlich in die eigene Tasche. Gluten ist ein signifikanter Auslöser bei psychiatrischen Erkrankungen wie Angstzustände, bipolare Störung, Depression und Schizophrenie. [Jackson 2011]
Einer der Gründe für die generelle, körperliche Müdigkeit kann auch eine Schilddrüsenunterfunktion sein. Diese und andere Krankheiten wie Anämie, Osteoporose, unerklärliche neurologische Syndrome, Unfruchtbarkeit, rheumatoide Arthritis, chronische Hypertransaminasämie unbekannter Ursache und Autoimmunerkrankungen wie autoimmune Schilddrüsenerkrankung, Typ 1 Diabetes mellitus, autoimmune Lebererkrankungen und entzündliche Darmerkrankungen treten bei Zöliakie gehäuft auf. [Ch'ng 2007], [Mainardi 2002], [Sategna-Guidetti 2001]
Weizenprotein (Gliadin/Gluten) aktiviert Zonulin, unabhängig von der genetischen Expression von Autoimmunität, was zu erhöhter Darmpermeabilität für Makromoleküle führt. [Drago 2006], [Lammers 2007] (Zonulin öffnet die Tight Junctions) Wer also über längere Zeit glutenhaltige Nahrung isst, obwohl er bereits unwissentlich eine Zöliakie oder Glutensensibilität entwickelt hat, der setzt sich einer sukzessiven Zerstörung seines Darms aus. Nervenzellen im Darm sind sogar ein primäres Ziel von Gliadin. Internisten prüfen leider nicht grundsätzlich auf Gliadin-Antikörper und das obwohl die Symptome im Darm weder einheitlich noch sonderlich stark ausgeprägt sein müssen.
Sie fragen sich, was hat Zöliakie/Glutensensibilität mit einer degenerativen Motorneuronerkrankung zu tun? Jetzt ist der Moment in dem Sie staunen werden. In einer Studie wurde bei einer Gruppe gesunder Patienten eine 12%ige Prävalenz von zirkulierenden Antigliadin-Antikörpern im Blut gemessen. 12/100 bzw. 1/8 ist hier jedoch ungewöhnlich hoch gegriffen, denn Untersuchungen zeigen, dass etwa nur 1/300 bis 1/85 von Zöliakie betroffen sind. [Lebenthal 2002] Bei den Patienten mit neurologischen Erkrankungen bekannter Ätiologie (Ursache) lag die Prävalenz nur bei 5%. Das ist logisch, denn wenn die Ursache bekannt ist, dann kann Zöliakie als Ursache so ziemlich ausgeschlossen werden, daher der niedrige Wert. Bei Patienten mit neurologischen Erkrankungen unbekannter Ätiologie hingegen lag die Prävalenz für Zöliakie bei 57%, also 5-mal so hoch als bei den gesunden Patienten (bzw. 50-mal höher relativ zum Bevölkerungsdurchschnitt). Die Zöliakie führte bei diesen Patienten zu immunologisch vermittelten Schäden von Kleinhirn, Rückenmark und peripheren Nerven. [Hadjivassiliou 1996] Solche extra-intestinalen Manifestationen werden auch bei chronisch entzündlichen Darmerkrankungen wie M. Crohn und Colitis ulcerosa häufig beobachtet. Microglia, die Immunzellen in Gehirn und Rückenmark, erzeugen pro-inflammatorische Zytokine die in großer Anzahl neurodegenerativ wirken. Bei der Entstehung von Alzheimer ist dieser Zusammenhang bereits 1994 dokumentiert worden [Akiyama 1994] und wird bis heute genauer untersucht [Wang 2015]. Es gibt tatsächlich keine einzige schwere Nervenkrankheit an der chronische Entzündungen nicht beteiligt sind. Bei der Degeneration von Motorneuronen wie bei der amyotrophen Lateralsklerose sind ebenso neuroinflammatorische Prozesse beteiligt. [McGeer 2002] Die Ursache scheint Ärzte jedoch nicht sonderlich zu interessieren.
Ein Patient bei dem ursprünglich geglaubt wurde, dass bei ihm eine Motorneuronkrankheit wie Amyotrophe Lateralsklerose vorliegt, bildeten sich die Symptome, die physikalischen- und Laborbefunde durch eine glutenfreie Diät binnen 23 Monate zurück. [Brown 2010]
Glutenataxie
2001 wurde die Glutenataxie bereits an der Universität Tübingen beschrieben. Glutensensibilität führt zu immunologisch bedingter Degeneration des Kleinhirns. In der Untersuchung waren 12/104 Ataxiepatienten Gluten-sensibel also 10 bis 30-mal so oft wie im Bevölkerungsdurchschnitt. Alle Patienten zeigten eine zerebelläre Atrophie im MRT. [Bürk 2001]Seit der Namensgebung der Amyotrophen Lateralsklerose vor über 150 Jahren hat es immerhin bis zum Jahr 2015 gedauert bis der Zusammenhang zwischen Gluten und Amyotrophe Lateralsklerose untersucht wurde: In bestimmten Fällen könnte ein Amyotrophe Lateralsklerose-Syndrom mit Autoimmunität und Glutensensitivität assoziiert sein. [Gadoth 2015] (Immerhin ist inzwischen die Rede von einem Syndrom) 2017 zeigte eine weitere Untersuchung keinen Zusammenhang. [Visser 2017] Getestet wurde in beiden Studien auf IgA Antikörper und genetische Zusammenhänge. Das ist jedoch die typische Vorgehensweise bei Zöliakie, für Glutensensitivität gibt es jedoch gar keine Untersuchungsmetoden. Natürlich sollte auf Zöliakie und auch auf Weizenallergie untersucht werden, allerdings sollte man hier keine positiven Befunde erwarten. Danach kann Glutensensitivität nur durch Glutenverzicht getestet werden.
Glutenempfindlichkeit & periphere Neuropathie
Periphere Neuropathie ist die zweithäufigste Manifestation der Glutenempfindlichkeit. In einer Studie von 101 Patienten mit idiopathischer (ohne bekannte Ursache) peripherer Neuropathie betrug die Anwesenheit der Glutenempfindlichkeit 40%. Die häufigste Art der peripheren Neuropathie war sensomotorische axonale (26) gefolgt von mononeuropathie multiplex (15), reine motorische Neuropathie (10), kleine Faserneuropathie (4) und gemischte axonale und demyelinisierung (2). Periphere Neuropathie ist mit einer chronischen und allmählichen Progression verbunden. [Hadjivassiliou 2002b]Bereits im Jahr 1966 veröffentlichte Cooke & Smith ein Übersichtsreferat über 16 erwachsene Patienten mit neurologischen Erkrankungen, die mit Zöliakie assoziiert waren. Zehn dieser Patienten hatten eine schwere progressive Neuropathie.
Zöliakie und Glutamin-Mangel
Wenn Amyotrophe Lateralsklerose Patienten lediglich an Muskelschwund leiden würden, dann müsste es auch nicht wenige deutlich übergewichtige Amyotrophe Lateralsklerose Patienten geben, diese gibt es aber nicht. Es handelt sich also eher um eine Kachexie, also eine krankhafte, sehr starke Abmagerung. Kachexie entsteht bei chronischen, auszehrenden Krankheiten, wie z.B. Krebs, AIDS, COPD, rheumatoide Arthritis, Diabetes Typ I, etc.. Es ist aber nicht so, als würde ein HIV-Virus die Muskeln wie Pac-Man auffressen. Bei AIDS entsteht der Gewebeverlust nicht durch den Virus sondern wahrscheinlich durch Glutamin-Mangel. [Shabert 1996] Glutamin-Mangel ist ein extremes Warnzeichen für eine schwere Stoffwechselstörung und eine immunologische Erkrankung. Es kann nicht sein, dass die Skeletmuskulatur kein Glutamin mehr speichert, weil anderenorts Nervenzellen absterben. Es ist viel mehr so, dass alle Zellen belastet sind. Es ist vollkommen ausgeschlossen, dass Nervenzellen das gesamte Glutamin aufbrauchen.Malabsorption und Kupfermangel
Zöliakie assoziierte Malabsorption kann zu Kupfermangel führen. Einige Forscher haben eine Rolle des langfristigen marginalen Kupfermangels in der Ätiologie verschiedener degenerativer Krankheiten vorgeschlagen. Marginaler Kupfermangel ist sehr schwierig serologisch zu untersuchen. [Bonham 2002] Beim Malabsorptionssyndrom wurde das jedoch genauer untersucht: Serumceruloplasmin betrug weniger als 5 mg / dl bei Normalwerten von 21-53 mg / dl und Serumkupfer betrug 12 mcg / dl (Normalwerte 85 bis 155 mcg / dl). [Hayton 1995]Glutamin- & Arginin-Mangel
Das Stresshormon Cortisol aktiviert die Bildung von Ornithin und Prolin aus Arginin und die Bildung von Glutamat, Alanin, Aspartat, Ornithin, Citrullin und Prolin aus Glutamin in Enterozyten (Resorpzionszellen im Darm). [Flynn 1997] Wohlgemerkt, Stress führt zur Bildung von Glutamat zu Lasten von Glutamin. Arginin-Mangel ist also typisch bei chronischem Stress und gilt daher als Stressmarker. Bei Arginin-Mangel wird vermehrt Glutamin in Glutamat umgewandelt, da Ornithin und Prolin zur Bildung von Glutamat fehlen. Arginin-Mangel verstärkt also den Glutamin-Mangel. Außerdem schwächt Arginin-Mangel den Harnstoffzyklus, sodass weniger Ammoniak neutralisiert wird. Was enthält unsere Knochenbrühe? Viel Kollagen mit 50% Prolin. Die Bildung von Prolin aus Arginin fällt damit flach und weniger Arginin wird verbraucht. Et voila! Eine Anti-Stress-Suppe.Studien haben gezeigt, dass chronischer Arginin-Mangel zu Degeneration von Gehirnzellen führt. Forscher deuten darauf hin, dass dieser Zustand durch eine Autoimmun-Reaktion verursacht wird, bei der das Enzym Arginase ungewöhnlich hohe Mengen Arginin abbaut. Im Tierversuch schützte die Hemmung dieses Prozesses z.B. vor Alzheimer. [Kan 2015] Also Stress oder eine Autoimmun-Reaktion führen zu Arginin-Mangel und Arginin-Mangel wirkt dann neurodegenerativ. Dieser Zusammenhang ist äußerst bemerkenswert, denn Stress aktiviert Leaky-Gut und Leaky-Gut wird als Ursache für Autoimmunkrankheiten beschrieben. [Fasano 2011], [Groschwitz 2009], [Fasano 2011b]
Außerdem ist Arginin als Vorläufer von Ornithin am Harnstoffzyklus beteiligt und spielt daher eine wichtige Rolle bei der Entgiftung von Ammoniak. [Brittenden 1994] Da auch Glutaminmangel, wie bereits erwähnt, zu einer Ammoniakbelastung führen kann und Arginin zur Entgiftung verbraucht wird, ist Arginin-Mangel auch ohne Autoimmun-Reaktion möglich.
Wenn Glutamin aufgebraucht wurde und auf Grund falscher Ernährung kein Glutamin mehr synthetisiert werden kann, droht eine Hyperammonämie. Sie fragen sich, was hat denn bitte Ammoniak mit Amyotrophe Lateralsklerose zu tun? Hyperammonämie gilt als mögliche Ursache von Amyotrophe Lateralsklerose. Der Fachbegriff ist Ammoniak Induzierte Amyotrophe Lateralsklerose. [Parekh 2015]
Im Tierversuch führte Cortisol zu Muskelatrophie, was durch Gabe von Glutamin um 50% gedämpft wurde. [Hickson 1995], [Hickson 1997]
Chronischer Stress macht das Gehirn empfänglicher für neurotoxische Belastungen von Nervenzellen.
Beim Menschen zeigte sich durch erhöhtes Cortisol im Plasma eine Steigerung der Glutaminfreisetzung um 40% (325 +/- 28 -> 453 +/- 28 mumol). [Darmaun 1988]
Hyperhomocysteinämie
Mehr als die Hälfte der über 50jährigen haben zu viel Homocystein im Blut. Möglichweise einer der Gründe, warum Menschen erst im mittleren Alter an Amyotrophe Lateralsklerose erkranken. Reine Spekulation?Homocystein gilt als neurotoxisch. [Kruman 2000]
Ein erhöhtes Gesamtplasma-Homocystein kann zu oxidativem Stress führen, was zu einer erhöhten Aktivität von antioxidativen Enzymen im Blutkreislauf führt. Homocystein reagiert vor allem mit Superoxid Dismutase und Glutathion Peroxidase. [Moat 2000] Homocystein muss daher als Dieb verstanden werden; er stiehlt intrazeluläres, antioxidatives Potential, welches dann für das Nervensystem nur noch vermindert zur Verfügung steht.
Homocystein wird z.B. mit Folsäure und Vitamin B12 behandelt:
Eine Metaanalyse wertete die Daten von 12 klinischen Studien mit insgesamt 1.114 Patienten aus. Die tägliche Einnahme von 0,5 - 5 mg Folsäure führte zu einer 25 %-igen Senkung des Gesamt-Homocysteinspiegels. Die erforderliche tägliche Minimaldosis zur Erreichung einer bestmöglichen Homocystein-Senkung lag bei ungefähr 0,5 mg. Der Effekt der Folsäuretherapie ist insbesondere von den Ausgangswerten des Homocysteins abhängig: je höher die Werte vor der Therapie waren, desto stärker die Homocystein-senkende Wirkung der Folsäure. Vitamin B12 (0,02-1 mg täglich) führte zu einer zusätzlichen Homocystein-Senkung von ca. 7 %. Zudem könnte die zusätzliche Vitamin B12-Gabe dem theoretischen Risiko einer Neuropathie vorbeugen, das bei Patienten mit Vitamin B12-Mangel unter Folsäure-Substitution besteht. Vitamin B6 (2 - 50 mg täglich) und Betaine (ein Methylgruppendonor, der am Methionin-Metabolismus beteiligt ist) waren deutlich weniger effektiv, und sind daher in der Regel verzichtbar. [Homocysteine Lowering Trialists' Collaboration 1998 ] Wer Vitamin B12-Mangel hat und Folsäure ohne B12 substituiert riskiert eine Neuropathie. Folsäure sollte daher immer zusammen mit Vitamin B12 eingenommen werden.
Orthomolekulare Lebensmittel für Folsäure & Vitamin B12: Hühner- und andere Geflügelleber
Das sind Tolle Ergebnisse, aber wir wollen mal nicht vergessen, dass Folsäure und Vitamin B12 in unserem Körper wichtigere Aufgaben haben, als sich mit Homocystein "herumzuschlagen". Besser wir beheben die Ursachen, dann können sich die Nutrienten um wichtigeres kümmern.
Das Homocystein ist beim Cushing-Syndrom erhöht. [Faggiano 2005], [Terzolo 2004] Damit ist spätestens jetzt klar, dass das Absenken des Cortisol-Spiegels eine der wichtigsten Therapieziele bei Amyotrophe Lateralsklerose ist. Interessiert das Neurologen? Nö.
Der Homocystein-Spiegel war bei Menschen mit sitzender Lebensweise um 10% höher. [Dankner 2005] Gute Freunde trifft man immer wieder. Ich sag immer, der optimale "Extremsport" für Patienten mit Amyotrophe Lateralsklerose heißt: Nicht-Sitzen. Bowling, Golf, Billiard, Darts, Kuscheln - alles wunderbar, aber mit "nicht lange sitzen" ist schon viel getan.
Vier Stunden nach einer Methionin-Belastung hatten an Cholin-Mangel erkrankte Männer Plasma-Homocystein-Konzentrationen, die 35% höher waren, als die von Männern die keinen Cholin-Mangel hatten. [da Costa 2005] Welches Lebensmittel hat am meisten Cholin? Das Hühnerei (Eigelb). Und wieder ein guter Grund mehr Eier zu essen.
Standartfrage: Was hat das mit Amyotrophe Lateralsklerose zu tun?
Die Bedeutung einer fehlerhaften Komplexierung von Kupfer, wird bei der vererblichen Form der Amyotrophen Lateralsklerose deutlich, bei der ein Enzymdefekt der Kupfer/Zink-Superoxid-Dismutase (Cu/Zn-SOD, SOD1) vorliegt, ein ubiquitär (überall) exprimiertes Enzym zum Abbau von Superoxidradikalen, welches ein Prozent des zellulären Gesamtproteins repräsentiert. Genau diese fehlerhafte Komplexierung von Kupfer kann auch durch Homocystein entstehen. [da Waggoner 1999] Was hier passiert ist folgendes: Normalerweise schützt Superoxid-Dismutase die Mitochondrien vor Superoxidradikalen, doch durch Homocystein entsteht genau das Gegenteil, es entstehen freie Radikale.
2. Standartfrage: Was hat das mit dem Darm zu tun?
Bei 62 Patienten mit chronisch entzündlichen Darmerkrankungen war die mittlere Plasmakonzentration von Homocystein höher als bei 183 gesunden Kontrollen (12.2 vs. 10.5 micromol/l). [Cattaneo 1998]
Die Ursache wird Sie nicht überraschen:
Eine Darmentzündung geht häufig mit einem Mangel an wichtigen Spurenelementen und Vitaminen einher. Patienten mit Darmentzündung sollten daher einmal jährlich ihren Serumspiegel von Zink, Eisen, dem Speichereiweiß Ferritin und von Vitamin B12 überprüfen lassen. Dies gelte auch für beschwerdefreie Patienten. Außerdem haben viele Erkrankte einen Mangel an Kalzium, Magnesium oder Folsäure. [Ärzte Zeitung] Darmentzündungen wiederum können verschiedene Ursachen haben, die sich aber im Gegensatz zu Amyotrophe Lateralsklerose gut behandeln lassen. Leider herrscht das Vorurteil, dass man bei einer Darmkrankheit extreme gastrointestinale Symptome haben muss, wie etwa Krämpfe oder starker Durchfall, aber das Gegenteil ist der Fall. Eine moderne Stuhl Untersuchung kann sehr gute Anhaltspunkte liefern. Darmentzündung⇒B12-Folsäure-Mangel⇒Hyperhomocysteinämie⇒Oxidativer Stress⇒Neurodegeneration. Darmentzündungen als Ursache von Amyotrophe Lateralsklerose? Ziemlich weit hergeholt? Wir wissen heute, dass erhöhter oxidativer Stress ein Teil dieser Multi-Systemerkrankung ist und die Möglichkeit, dass Verdauung und Ernährungsweise bei oxidativem Stress keine Rolle spielt, halte ich für so gut wie ausgeschlossen.
Aber warum haben so viele ältere Menschen so hohe Homocystein-Werte? Und wieder liegt die Antwort im Verdauungstrakt:
Ungefähr 50 Prozent aller Menschen über 65 Jahre entwickeln eine Zellschwäche an der Magenwand, die die Sekretion und Funktion des Magens beeinträchtigt. Die verminderte Sekretion des Intrinsic-Faktors wiederum verschlechtert die Bioverfügbarkeit von Vitamin B12 erheblich und führt bei vielen Menschen zu einem Mangelzustand. [Lindenbaum 1994]
Neurologen die den Vitamin-B12-Status nicht abklären, handeln in meinen Augen fahrlässig. Diesen Eindruck bekommt man, wenn man sich die typischen Mangelsymptome anschaut:
Zu den möglichen Symptomen gehören Neuropathie, Versagen des Positionssinns und der Vibrationsempfindung, Schwäche der Beine, verminderter Geschmack, vorzeitiges Ergrauen der Haare, rote Zunge, Erschöpfung, verminderte Fähigkeit Farben mit leicht unterschiedlichen Farbtönen zuzuordnen und Gedächtnisschwäche. [Fine 1992.]
Aber selbst wenn Ihr Arzt Vitamin B12 im Serum überprüft, das Resultat könnte im Normalbereich liegen und trotzdem könnten Sie von einem höheren B12 Serum-Spiegel profitieren, weil Sie einen höheren Bedarf haben als gesunde Menschen. Das nennt man biochemische Individualität. Und genau das bestätigte eine Untersuchung:
37 Patienten profitierten von einer hämatologischen oder neurologischen Verbesserung durch eine Vitamin B12-Therapie. Wenn die Behandlung nur auf Patienten mit niedrigen oder mittleren Vitamin-B12-Spiegeln und erhöhten Methylmalonsäure- und Homocysteinspiegeln beschränkt gewesen wäre, wären 63% der Responder nicht behandelt worden. Umgekehrt hatten von 25 Patienten, die nicht auf eine Vitamin B12-Therapie ansprachen, 45% ein niedriges Serum-Vitamin B12, 45% eine erhöhte Methylmalonsäure und 43% ein hohes Homocystein. [Solomon 2005] Also das ist doch Biochemie wie sie lacht und lebt, oder? Normbereiche für die Katz! Patienten mit serologisch nachgewiesenem Vitamin B12-Mangel profitierten nicht von der Vitamin B12-Therapie und Patienten mit serologisch normalen Vitamin B12-Werten profitierten. Die Plasma-Homocystein-Konzentration ist der zuverlässigste Marker für Vitamin B12 - das müssen Sie sowieso überprüfen lassen, aber egal was auch immer dabei herauskommt, es gibt nur eine Möglichkeit herauszufinden ob Sie von Vitamin B12 profitieren können: Gehen Sie zu Ihrem Hausarzt und lassen Sie sich Vitamin B12 intramuskulär Spritzen und beobachten Sie, ob es Ihnen damit besser geht. Wenn ja, weiter Spritzen!
Obwohl der Vitamin-B12-Wert im Blut normal oder sogar erhöht ist, kann auf zellulärer Ebene ein B12-Mangel vorliegen. Bei Schwermetallbelastungen (Quecksilber, Blei) kann das normale Vitamin B12 (Cyanocobalamin) nicht in das besonders in den Nervenzellen benötigte Methyl-Cobalamin umgewandelt werden.
Studien an gesunden Probanden zeigen, dass 6 g / Tag Betain die Plasma-Homocystein-Konzentrationen um 5% bis 20% senken kann. 4 g / Tag senkte die Plasma-Homocystein-Konzentrationen um 1.23 µmol/L. [da McRae 2013] Welches Lebensmittel hat am meisten Betain? Quinoa hat immerhin ca. 1 g / Portion und sollte daher immer auf Ihrem Einkaufszettel stehen.
Anabolismus
Amyotrophe Lateralsklerose und Anabolika hört sich an wie ein schlechter Witz, weil man Anabolika eher mit größenwahnsinnigen Extremsportlern assoziiert, doch das ist genau die falsche Denkweise. Ohne anabolen Stoffwechsel würden ansonsten völlig gesunde Menschen binnen kürzester Zeit muskulär vollständig atrophieren. Es wäre also mehr als logisch Störungen des anabolen Stoffwechsels als Ursache zu vermuten, trotzdem hat die medizinische Forschung das Thema Anabolismus bei Amyotrophe Lateralsklerose seit je her ignoriert. Das lag natürlich an dem Fehlglauben, dass Amyotrophe Lateralsklerose eine reine Nervenkrankheit sei. Ich gehe sogar davon aus, dass das Versagen des anabolen Stoffwechsels eine zentrale Rolle bei der amyotropher Lateralsklerose spielt.Ground Zero
Eine banale Frage die sich offensichtlich zu wenige Forscher stellen, ist, warum bei amyotropher Lateralsklerose nur die motorischen Nerven leiden, aber nicht die sensorischen. Die Antwort liegt auf der Hand: Amyotrophe Lateralsklerose muss etwas mit dem Stoffwechsel der Muskulatur zu tun haben, denn sensorische Nerven sind sehr ähnlich wie die motorischen, außer, dass sie an ihren Enden nicht in einer Muskelfaser verwurzelt sind. Das neurologische Ground Zero liegt also nicht im zentralen Nervensystem, sondern eher am Ende der motorischen Nervenbahnen - in der Muskulatur. Genau diese Theorie haben Forscher vom Würzburger Universitätsklinikum jüngst bestätigt. Sie schalteten im Mausmodel ein Gen aus, welches an den Synapsen den Abbau der Vesikel steuert, die mit dem Botenstoff Acetylcholin gefüllt sind und die Erregung vom Nerven zu den Muskeln transportieren. Die Inaktivierung des Gens führte zu einer Motoneuron-Erkrankung. [Lüningschrör 2016] D.h. dass Schäden an der Verbindungsstelle zwischen Nerv und Muskelphaser ein substanzieller Teil der Ursache von Amyotrophe Lateralsklerose sein kann. Das Hauptproblem bei Amyotrophe Lateralsklerose ist also möglicherweise eher der Muskelstoffwechsel und die Nervenverbindungen direkt am Muskel sind nur Teil eines muskulären Kollateralschadens. Das sind gute Nachrichten, denn im Gegensatz zum Nerv, hat der Muskel sehr gute Regenerationsmöglichkeiten und selbstverständlich spielt die Ernährung dabei eine entscheidende Rolle.Brain-derived neurotrophic factor (BDNF)
Nach dem Stress kam die Depression. Depression ist eigentlich keine psychisch bedingte Krankheit, sondern eine stressbedingte, neurodegenerative Erkrankung des Hypocampus. Die zukünftigen Antidepressiva werden Antistress Medikamente sein. Eine der Folgen von Depression ist die Dysregulation der BDNF-Expression. BDNF wirkt auf verschiedene Neuronen des zentralen und des peripheren Nervensystems. Es wirkt beim Schutz existierender Nervenzellen und Synapsen mit und fördert das Wachstum neuer. [Acheson 1995]BDNF macht sich auch im neuromuskulären System nützlich, wo es Neuromotoren vor Degegeneation schützt. Der Neuromotor ist das wichtigste Element im Muskel. Ohne den Neuromotor ist der Muskel wie ein Motor ohne Zündung. Neuromotorische Degradation ist z.B. auch beteiligt an der altersbedingten Muskelatrophie. BDNF hilft im wahrsten Sinne des Wortes dabei, die Degeneration des Gehirns zu verhindern und sogar umzukehren, ebenso wie es die altersbedingte Muskelatrophie verhindert und rückgängig macht. Grund genug BDNF unter die Lupe zu nehmen.
Man will es kaum glauben, aber eine bakterielle Fehlbesiedlung des Darms verändern den BDNF Spiegel im Gehirn. Wissenschaftler veränderten die Zusammensetzung und Besiedlungsdichte der Darmbakterien bei Mäusen durch die Gabe von Antibiotika. Anschließende Verhaltensanalysen der Versuchstiere zeigten charakteristische Veränderungen: Manche Nager wurden waghalsiger, andere dagegen ängstlicher, als sie es vor der Umgestaltung ihrer Darmflora gewesen waren. Diese Effekte wurden von einem veränderten Spiegel des Botenstoffes BDNF im Gehirn begleitet. Von dieser Substanz ist bereits bekannt, dass sie im Zusammenhang mit Depressionen und Angstzuständen steht. [Bercik 2009]
Eine gesunde Darmflora fördert eine gesunde Psyche möglicherweise durch optimale BDNF Spiegel im Gehirn. Eine gestörte Darmflora ist somit problematisch. Über die Ernährung können wir jedoch erheblichen Einfluss auf unsere Darmgesundheit nehmen, im positiven wie im negativen Sinne.
Entspanntes Gehen (bzw. nicht sitzen) ist die einzige Sportart die ich für Amyotrophe Lateralsklerose Patienten empfehle. Bewegung hilft den Körper von Stress-Chemikalien zu befreien, welche zu Depressionen führen, depressive Perioden wiederum lassen den Hippocampus schrumpfen. Bewegung hingegeben erhöht das Volumen der grauen Substanz im Hippocampus. Bewegung fördert die Neurogenese, d. h. die Fähigkeit des Gehirns sich durch Wachstum neuer Gehirnzellen anzupassen. [Erickson 2011]
Zucker und gesättigte Fette hingegen unterdrücken den vom Hirn abgeleiteten neurotrophen Faktor (BDNF). [Wu 2004], [Molteni 2002]
Körperliche Aktivität (bei Amyotropher Lateralsklerose nur ohne Krafteinsatz) erhöht die BDNF-Expression. [Schaaf 2000]
Cortisol unterdrückt die BDNF-Expression. [Griesbach 2004]
Und Omega-3-Fettsäuren sind auch wieder mit im Team als neuroprotektiver und neurogener Wirkstoff. Es erhöht die BDNF-Synthese und die intrazelluläre Signalübertragung in Neuronen. [Balanzá-Martínez 2011]
Ein Forscherteam entdeckte, dass die Gabe von Blaubeerpulver bei Mäusen zu einem erhöhten Spiegel des Nervensignalstoffes BDNF (brain-derived neurotrophic factor) im Hippocampus führt und dadurch dort das Wachstum neuer Nervenzellen fördert. In Gedächtnistests erzielten die Mäuse 30 Prozent bessere Ergebnisse als ihre herkömmlich gefütterten Artgenossen. [Rendeiro 2012] Nur eine Tierstudie? Es gibt zahlreiche positive Studien mit Blaubeeren bei allen neurologischen Krankheiten, überwiegend als Antioxidant im Gehirn, doch ein Zusammenhang mit BDNF ist beeindruckend und ist daher besonders hervorzuheben.
Das Gleiche gilt auch für Kurkuma - viele positive Studien und BDNF-Aktiv im Tierversuch: Die Supplementierung von Curcumin in der Nahrung reduzierte die oxidative Schädigung drastisch und normalisierte die Gehalte an BDNF-Synapsin I und CREB, die nach einer künstlich provozierten Gehirnerschütterung bei Ratten verändert worden waren. Darüber hinaus wirkte Curcumin der kognitiven Beeinträchtigung entgegen. [Wu 2006]
Wachstumsfaktor BDNF fördert das neuronale Wachstum und das neuronale Überleben und seine Freisetzung ist Abhängig von glutamatergischer synaptischer Aktivität. [Carvalho 2008], [Lu 2003] Die BDNF-Expression wird durch Stress reduziert und z.B. durch Meditation erhöht. Aktive Entspannungstechniken können also die neuronale Regeneration fördern und den Stressreaktionen im Körper entgegenwirken. Wenn Amyotrophe Lateralsklerose Patienten trainieren, dann trainieren sie ncht die Muskeln sondern entspannen den gesamten Körper und Geist. Asana-Yoga, Stressmanagement, Lachen, Ausreichend Schlaf (min. 7 Stunden) oder die finanzielle Situation verbessern sind wirksame Gegenmaßnahmen bei stressbedingter Hypercortisolämie. Wenn die BDNF-Expression Glutamat verbraucht, dann eine geringe BDNF-Expression möglicherweise auch der Grund für erhöhte Glutamat-Spiegel bei Patienten mit Amyotrophe Lateralsklerose. Hierin könnte auch eine Begründung für die geringe Effektivität von Glutamat-Antagonisten liegen. Glutamat zu blockieren erscheint wenig Sinnvoll, denn Glutamat soll ja schließlich die Transkription und Freisetzung von BDNF erhöhen. Entspannen, Sorgen eliminieren, Ängste abbauen und jegliche Formen von Meditation gibt dem Glutamat also seine natürliche Aufgabe zurück.
Beim Training, Fasten und beim Saunabaden setzen Nervenzellen Proteine frei, die als neurotrophe Faktoren bekannt sind, wie z.B. BDNF, welche Hirnstammzellen aktiviert, um neue Neuronen zu produzieren.
Interleukin-6 (IL-6)
Interleukin-6 (IL-6), ein entzündliches Zytokin, ist stark mit funktionellem Abfall, Kraftverlust, Sarkopenie (Abbau der Skelettmuskulatur) und Mortalität verbunden. [Jensen 2008]Wenn Sie Ihre eigenen Untersuchungen in Auftrag geben und dann sowas im Blutbild finden, dann wissen Sie, dass jemand seinen Job nicht macht:
Chronischer Stress führt zu 4-mal höheren Interleukin-6-Serum-Werten. [Kiecolt-Glaser 2010] Interleukin-6 wird unter anderem in der Darmmukosa und im Muskel gebildet.
Die Behandlung von normalen Mäusen mit Interleukin-6 (IL-6) führte zu einer signifikante Abnahme der hepatischen Sulfatspiegel und eine Erhöhung des Harnstoff / Glutamin-Verhältnisses innerhalb von 4 h. [Hack 1996] Interleukin-6⇒Glutamin-Mangel
Alle wichtigen degenerativen Erkrankungen sind mit einer Mikrogliaaktivierung und der übermäßigen Produktion von Zytokinen wie Interleukin-1 beta, Interleukin-6 und Tumor-Nekrose-Faktor-alpha assoziiert. [Blaylock 2002]
Sie fragen sich: "OK, Interleukin-6 ist ein Problem, aber ist das denn nie bei Amyotrophe Lateralsklerose überprüft worden?". Na klar doch! 2006 wurde das bereits untersucht und 2017 in einer Meta-Analyse sogar schön zusammengefasst worden: Interleukin-6 und diverse andere Zytokine waren bei Patienten mit Amyotropher Lateralsklerose signifikant erhöht. [Hu 2017] Tolle Ergebnisse, doch leider ist klinische Wissenschaft nicht dasselbe wie klinische Praxis. Bevor es klinische Studien in die Leitlinien zur Behandlung von amyotrophe Lateralsklerose schaffen, vergehen viele Jahrzehnte und das Meiste wird niemals im Alltag der Neurologen auftauchen. Vieles was Wissenschaftler erforscht haben ist in der klinischen Praxis sogar regelrecht inexistent. Damit ein Entzündungshemmer zugelassen wird muss sowieso erstmal ein Pharmaunternehmen sich der Sache annehmen. Ob das jemals passieren wird ist jedoch äußerst fraglich und wenn, dann behebt das weder die Ursache der Entzündung noch die diversen anderen Gesundheitsprobleme von Amyotrophe Lateralsklerose Patienten. Eine klinische Studie wird dann eine Monotherapie mit Placebo vergleichen und keine signifikanten Ergebnisse hervorbringen.
Die Immunologie schaut dann gerne mal so aus: Das 15-16-fache vom Mittelwert bzw. das 8-fache vom Maximalwert:
Es gibt Studien die IL-6 als signifikant erhöht belegten, andere Studien zeigen keine Signifikanz. Mann sollte aber auch nicht erwarten, dass, alle genau dasselbe Syndrom haben, denn wahrscheinlich gibt es verschiedene Amyotrophe Lateralsklerose-Typen mit unterschiedlicher multisystemischer Pathogenese.
Forscher der Harvard Medical School in Boston haben mittels 18F-fluorodexoyglucose PET / CT und einem radioaktiv markierten Glukoseanalogon untersucht, was bei Stress passiert. Sie stellten fest, dass es in Stresssituationen über Aktivierung der Amygdala zu einer Reizweiterleitung kommt, die im Knochenmark zur erhöhten Aktivität und Freisetzung von pro-inflammatorischen Zytokinen führt. [Tawakol 2016] Stress = Entzündung.
Zu allem Überfluss neben AIDS, Krebs und Amyotrophe Lateralsklerose - was glauben Sie ist der Grund für Gebrechlichkeit im hohen Alter? Altersschwäche etwa? Weit gefehlt! erhöhte entzündliche Aktivität mit erhöhtem interleukin-6 (IL-6) und C-reactive protein (CRP). [Velissaris 2017] Wohl bemerkt, Gebrechlichkeit bedeutet primär neuromuskuläre Schwäche.
Metabolische Azidose
Stress macht uns sauer. Schuld daran ist die Überreizung des vegetativen Nervensystems in Kombination mit flacher Atmung, hochkalorischer Nahrung und Bewegungsmangel. Die Psyche beeinflusst direkt unsere Atmung, denn durch Anspannung und Ängste atmen wir flach. Unser Körper wird nicht mehr ausreichend mit Sauerstoff versorgt und atmet die Säuren in Form von "Kohlendioxid" nicht vollständig ab. Außerdem werden bei Stress vermehrt die Hormone Adrenalin und Noradrenalin ausgeschüttet, bei deren Verarbeitung entstehen Säuren, die zu einer Übersäuerung beitragen. Die Problematik ist hinreichend bekannt von der Übersäuerung bei kalorienreduzierten Diäten. Die Ursache sind auch hier hohe Stresshormone. Gougeon-Reyburn, Rejeanne, Ph.D., et al, American Journal of Medical Sciences, August 1991;302(2):67-73.Glutamin wird bei Übersäuerung zu Basen reduziert (über α-Ketoglutarate zu Bikarbonat) und dient so als Puffer für diätische Säuren. Um aus der diätischen Glutaminsäure Glutamin zu bilden wird basisches Ammoniak benötigt. Ammoniak wird jedoch in den Nieren zu Neutralisation von Säuren genutzt. Dieser äußerst gravierende Zusammenhang ist bereits seit den 60er Jahren bekannt. [OWEN 1963] Die Azidose ist also ein Art Dieb der Ammoniak entwendet, sodass die Glutaminsäure nicht zum Zug kommt. So entsteht der zu hohe Glutamat-Spiegel bei gleichzeitigem Glutamin-Mangel.
Muskelschwund ist eine adaptive Reaktion auf Azidose [Williams 1991], [May 1986], [Cersosimo 1986], [Guder 1987]. Beim Muskelabbau werden Aminosäuren in den Blutkreislauf freigesetzt. Diese Aminosäuren liefern ein Substrat für die Lebersynthese von Glutamin. Glutamin wird von der Niere zur Synthese von Ammoniak [Owen 1963] verwendet. Ammoniakmoleküle akzeptieren spontan Protonen und werden als Ammoniumionen ausgeschieden; Die Ausscheidung von Ammonium entfernt so Protonen und vermindert die Azidose [Dawson-Hughes 2008]. Übersäuerung fördert also auch den katabolen Stoffwechsel der Muskulatur.
Metabolische Azidose kann den Muskel-Protein-Abbau verstärken. [Garibotto 1996]
Eine Übersäuerung des Darms führt zu einer Schädigung der Darmschleimhaut. Dadurch vermindert sich die Bildung von Lymphozyten. Folglich kommt es zu einem erhöhten Infektionsrisiko.
Beispielgebend für den Interorganstoffwechsel des Glutamins wurde die Stoffwechselsituation der Niere bei Azidose beschrieben. Welbourne hat in experimentellen Untersuchungen gezeigt, dass im Zustand der Azidose alle Organe dazu beitragen, dass der Glutaminfluss in Richtung Niere stimuliert wird, wo Glutamin zur Pufferung saurer Valenzen verwendet wird. Die Regulation dieses Stoffwechselkreises läuft primär über Glukokortikoide, die die Glutaminfreisetzung des Skelettmuskels stimulieren. Glutamin dient in der Niere zur Pufferung der anfallenden Hydroniumionen. Beim azidotischen Intensivpatienten hat Glutamin somit eine enorme Wichtigkeit als endogener Puffer. Eine Verabreichung von Endotoxin bewirkt eine verringerte Glutaminextraktion der Niere, die möglicherweise für die Beeinträchtigung des Säure/Basenhaushaltes im septischen Zustandsbild verantwortlich ist. [Welbourne 1987]
Azidose hemmt die Harnstoffsynthese in der Leber, die den auszuscheidenden Stickstoff dann in Form von Glutamin zur Niere schickt.
Im Detail: Glutamin ist für die Regulation des Säure-Basen-Haushalts von Bedeutung, weil es von den Nieren zur Bildung und Ausscheidung von sauren Ammoniumionen abgebaut wird. Während einer metabolischen Azidose steigt die Ausscheidung von Ammoniumionen um ein Mehrfaches an. Da Glutamin zwei Drittel der Ammoniumionen stellt, kommt es auch zu einem sehr starken Anstieg der Glutaminaufnahme durch die Nieren. Glutamin wird dabei in alpha-Ketoglutarat umgewandelt und von dort gibt es dann nur noch einen Weg: Glutamat. Eine metabolische Azidose führt daher zur Synthese von Glutamat auf Kosten von Glutamin, also genau das Gegenteil von dem was wir wollen.
Eine säure-arme Ernährung mit viel Obst, Gemüse, Nüssen und Hüsenfrüchten ist daher sehr wichtig um das ungünstige Glutamin/Glutamat-Verhältnis umzudrehen. Eine Ernährungsumstellung ist langfristig nützlicher als die vorübergehende Einnahme von Nahrungsergänzungsmitteln. Natürlich kann man eine basische Ernährung genauso gut mit Glutamin ergänzen um basisches Bikarbonat einzusparen.
Eine metabolische Azidose beschleunigt den Proteinkatabolismus der Skelettmuskulatur. Möglicherweise als Teil einer koordinierten homöostatischen Multi-Organ-Reaktion auf Azidose. Skelettmuskulatur und herabgesetzte Harnstoff-Produktion liefern den für die Nieren-Ammoniakese benötigten Stickstoff. [Williams 1991]
Kaliummangel / Hypokaliämie
Die wichtigste Base ist das Mineral Kalium. Hypokalämie bezieht sich auf niedrige Kaliumspiegel im Blut. Essentielle Nährstoffe wie Kalium helfen, die Nerven- und Muskelfunktion zu erhalten. Das meiste Kalium befindet sich im Muskel. Niedrige Kaliummengen im Blut können zu lebensbedrohlichen Bedingungen führen. [Burnell 1974]In der westlichen Ernährung ist Kaliummangel weit verbreitet. Hypokaliämie läßt sich jedoch nicht immer allein auf eine ungesunde Ernährung zurückführen. Eine der Ursachen für Hypokalämie ist das Cushing-Syndrom oder Perioden von starkem Stress. [Sharma 2015], [Veltri 2015] Eine milde Hypokaliämie führt bereits zu Krämpfen, Unwohlsein, Muskelschmerzen und Schwäche, bei starker Hypokalämie drohen Lähmungserscheinungen.
Kaliummangel führt zu Verringerung der Insulinausschüttung durch die beta-Zellen der Bauchspeicheldrüse. Hypoinsulinämie führt dann zu Hyperglykämien, was wiederum zu peripherer Neuropathie führen kann. Die wichtigste anabole Wirkung von Insulin auf die Skelettmuskulatur sind auf seine hemmende Wirkung auf den Proteinabbau zurückzuführen. Fehlet Insulin kann der Proteinabbau nicht verhindert werden - Muskelschwund droht. Da die Glukose nicht in die Muskelzelle transportiert wird entsteht Kraftverlust und eine schnelle körperliche Ermüdung (Ganzkörperschwächung). Dadurch wird der Körper weniger bewegt, was wiederum zu psychischem Stress, Übersäuerung, Darmträgheit und weiterer Schwächung der Muskulatur führt. Insulin wird auch für die Kaliumabsorption der Muskelzellen benötigt. Fehlt Insulin schafft es das ohnehin wenige Kalium noch nicht mal in die Muskelzellen. Daher sollte Kalium auch grundsätzlich im Plasma gemessen werden. [Stone 2016]
Vitamin D Mangel
Vitamin D Mangel ist mit einer schlechteren Prognose bei Amyotrophe Lateralsklerose assoziiert. [Wang 2016]Ein schwerer Vitamin-D-Mangel beschleunigt die Neurodegeneration um das 4-fache und ist mit einer deutlich kürzeren Lebenserwartung verbunden. [Camu 2013]

[Abu-Qare 2001] Abu-Qare AW, Abou-Donia MB. J Toxicol Environ Health B Crit Rev. 2001 Aug;4(3):313-32.
[Acheson 1995] Acheson A, et al. Nature. 1995 Mrz;374(6521):450-3.
[Aggarwal 2009] Aggarwal BB, Harikumar KB. Int J Biochem Cell Biol. 2009 Jan;41(1):40-59
[Akiyama 1994] Akiyama H. Tohoku J Exp Med. 1994 Nov; 174(3):295-303.
[Albrecht 2006] Albrecht J, Sonnewald U, Waagepetersen HS, Schousboe A. Front Biosci. 2006 Nov;12:332-43.
[Aldridge 2015] Aldridge DR, Tranah EJ, Shawcross DL. J Clin Exp Hepatol. 2015 Mrz;5(Suppl 1):S7-S20.
[Aleo 1985] Aleo JJ, Padh H. Proc Soc Exp Biol Med 179(1):128—31, 1985
[Alverdy 1990] Alverdy JC. JPEN J Parenter Enteral Nutr. 1990 Jul;14(4 Suppl):109S-113S.
[Amar 2008] Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, Chamontin B, Ferriéres J. Am J Clin Nutr. 2008 Mai;87(5):1219-23.
[Andreasen 2008] Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Møller K. Curr Med Chem. 2008 Aug;15(17):1697-705.
[Andrikopoulos 2016] Andrikopoulos S, et al. University of Melbourne. 07 Feb 2016
[Atassi 2011] Atassi N, Cook A, Pineda CM, Yerramilli-Rao P, Pulley D, Cudkowicz M. Amyotroph Lateral Scler. 2011 Mrz;12(2):109-12.
[Apostolakis 1989] Apostolakis M, Anogianakis G, Kallaras C, Zaraboukas T, Liangouris J, Nowack-Apostolaki E, Economou L. Brain Res Bull. 1989 Sep;23(3):257-62.
[Appel 1993] Appel SH, Smith RG, Engelhardt JI, Stefani E. J Neurol Sci. 1993 Sep;118(2):169-74.
[Appel 1994] Appel SH, Smith RG, Engelhardt JI, Stefani E. J Neurol Sci. 1994 Jul;124 Suppl:14-9.
[Aretaeus 1956] Aretaeus. Liber IV In: Corpus Medicorum Graecorum. Berlin: Akademie-Verlag GmbH 1956:74.
[Armstrong 2002] Armstrong LE, VanHeest JL. Sports Med. 2002 Aug;32(3):185-209.
[Armstrong 2007] Armstrong ML, Dua AK, Murrant CL. J Physiol. 2007 Jun;581(Pt 2):841-52.
[Arwert 2003] Arwert LI, Deijen JB, Drent ML. Nutr Neurosci. 2003 Nov;6(5):269-75.
[Arvanitakis 2004] Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Arch Neurol. 2004 Mai;61(5):661-6.
[Atkinson 2001] Atkinson KJ, Rao RK. Am J Physiol Gastrointest Liver Physiol. 2001 Mai;280(6):G1280-8.
[Aun 2009] Aun MV, Ribeiro MR, Costa Garcia CL, Agondi RC, Kalil J, Giavina-Bianchi P. J Asthma. 2009 Mai;46(4):399-401.
[Babu 2007] Babu GN, Kumar A, Chandra R, Puri SK, Kalita J, Misra UK. Neurochem Res. 2007 Sep;33(6):1145-9.
[Balanzá-Martínez 2008] Balanzá-Martínez V, et al. Expert Rev Neurother. 2011 Jul;11(7):1029-47.
[Barbeau 1974] Barbeau A., Preliminary study of glycine administration in patients with spasticity. Neurology 24:392, 1974.
[Barnes 2009] Barnes PJ, Adcock IM. Lancet. 2009 Jun;373(9678):1905-17.
[Black 1970] Black JT. J Neurol Neurosurg Psychiatry. 1970 Dez;33(6):864-70.
[Bailey 2013] Bailey MT, Lubach GR, Coe CL. J Pediatr Gastroenterol Nutr. 2004 Apr;38(4):414-21.
[Bala 2014] Bala S, Marcos M, Gattu A, Catalano D, Szabo G. PLoS One. 2014 Feb;9(5):e96864.
[Barbagallo Sangiorgi 1984] Barbagallo Sangiorgi G, Costanza G, Di Sciacca A, Frada' G, Cupidi G, Malta R, Affronti M, Barbagallo M. Age Ageing. 1984 Sep;13(5):309-12.
[Baydas 2002] Baydas G, Kutlu S, Naziroglu M, Canpolat S, Sandal S, Ozcan M, Kelestimur H. J Pineal Res. 2002 Dez;34(1):36-9.
[Bayer 2015] Wolfgang Bayer, Karlheinz Schmidt. Zs.f.Orthomol.Med. 2015; 2(02): 6-9. DOI: 10.1055/s-0035-1547545
[Baker 2010] Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S. Arch Neurol. 2010 Sep;68(1):51-7.
[Bennett 2008] Bennett MP, Lengacher C. Evid Based Complement Alternat Med. 2008 Mrz;5(1):37-40.
[Benjamin 2011] Benjamin J, Makharia G, Ahuja V, Anand Rajan KD, Kalaivani M, Gupta SD, Joshi YK. Dig Dis Sci. 2011 Mai;57(4):1000-12.
[Bercik 2009] Bercik P, et al. Gastroenterology. 2009 Aug;139(6):2102-2112.e1.
[Berk 1989] Berk LS, Tan SA, Fry WF, Napier BJ, Lee JW, Hubbard RW, Lewis JE, Eby WC. Am J Med Sci. 1989 Dez;298(6):390-6.
[Bergström 1967] Bergström J, Hultman E. Scand J Clin Lab Invest. 1967 Sep;19(3):218-28.
[Beydoun 2013] Beydoun MA, Beydoun HA, Shroff MR, Kitner-Triolo MH, Zonderman AB. Psychosom Med. 2013 Mai;75(5):486-96.
[Biolo 2004] Biolo G, Zorat F, Antonione R, Ciocchi B. Int J Biochem Cell Biol. 2004 Okt;37(10):2169-79.
[Black 1970] Black JT. J Neurol Neurosurg Psychiatry. 1970 Dez;33(6):864-70.
[Blaylock 2002] Blaylock RL, JANA, Winter 2002;5(1):15-32.
[BLC] Bundesverband der Lebensmittelchemiker/-innen im öffentlichen Dienst e.V. (BLC)
[Bonham 2002] Bonham M, O'Connor JM, Hannigan BM, Strain JJ. Br J Nutr. 2002 Mai;87(5):393-403.
[Bounous 1999] Bounous G, Molson J. Med Hypotheses. 1999 Dez;53(4):347-9.
[Borel 1998] Borel MJ, Williams PE, Jabbour K, Levenhagen D, Kaizer E, Flakoll PJ. JPEN J Parenter Enteral Nutr. 1998 Sep;22(5):280-5.
[Brenner 2017] Brenner SR. Med Hypotheses. 2017 Mai;102:89-90.
[Briese 2018] Briese M, et al. Proc Natl Acad Sci U S A. 2018 Mrz;115(12):E2859-E2868.
[Brillon 1995] Brillon DJ, Zheng B, Campbell RG, Matthews DE. Am J Physiol. 1995 Mrz;268(3 Pt 1):E501-13.
[Brittenden 1994] Brittenden J, Heys SD, Ross J, Park KG, Eremin O. Clin Sci (Lond). 1994 Feb;86(2):123-32.
[Brown 1908] Brown C W. Sprue and its treatment. London: J Bale, sons, and Danielson, 1908
[Brown 2010] Brown KJ, Jewells V, Herfarth H, Castillo M. AJNR Am J Neuroradiol. 2009 Nov;31(5):880-1.
[Bürk 2001] Bürk K, Bösch S, Müller CA, Melms A, Zühlke C, Stern M, Besenthal I, Skalej M, Ruck P, Ferber S, Klockgether T, Dichgans J. Brain. 2001 Mai;124(Pt 5):1013-9.
[Burks 2012] Burks AW, Tang M, Sicherer S, et al. ICON: Food allergy. J Allergy Clin Immunol 2012, 129:906-20
[Burton 1994] Burton; Helmut Rennke (1994). Renal Pathophysiology. Baltimore: Williams & Wilkins. ISBN 0-683-07354-0.
[Burnell 1974] Burnell JM, Teubner EJ, Simpson DP. Am J Physiol. 1974 Aug; 227(2):329-33
[Camu 2013] Camu W, Tremblier B, Plassot C, Alphandery S, Salsac C, Pageot N, Juntas-Morales R, Scamps F, Daures JP, Raoul C. Neurobiol Aging. 2013 Apr;35(5):1198-205.
[Candelario 2015] Candelario M, et al. J Ethnopharmacol. 2015 Feb;171:264-72.
[Cao 1998] Cao Y, Feng Z, Hoos A, Klimberg VS. JPEN J Parenter Enteral Nutr. 1998 Jul;22(4):224-7.
[Carmeli 2012] Carmeli C, Knyazeva MG, Cuénod M, Do KQ. PLoS One. 2012 Aug;7(2):e29341.
[Carroccio 1991] Carroccio A, Iacono G, Montalto G, Cavataio F, Di Marco C, Balsamo V, Notarbartolo A. Gut. 1991 Jul;32(7):796-9.
[Cattaneo 1998] Cattaneo M, et al. Thromb Haemost. 1998 Okt;80(4):542-5.
[Cersosimo 1986] Cersosimo E, Williams PE, Radosevich PM, Hoxworth BT, Lacy WW, Abumrad NN. Am J Physiol. 1986 Jun;250(6 Pt 1):E622-8.
[Cerra 1991] Cerra FB. Am J Surg. 1991 Feb;161(2):230-4.
[Chapman 1970] Chapman LF. Q J Stud Alcohol. 1970 May; 5:Suppl 5:67-86
[Chastain 2012] Chastain EM, Miller SD (2012). "Molecular mimicry as an inducing trigger for CNS autoimmune demyelinating disease". Immunol. Rev. 245 (1): 227–38. doi:10.1111/j.1600-065X.2011.01076.x.
[Chen 2017] Chen C, Nakagawa S, An Y, Ito K, Kitaichi Y, Kusumi I. Front Neuroendocrinol. 2017 Jan;44:83-102.
[Ch'ng 2007] Ch'ng CL, Jones MK, Kingham JG. Clin Med Res. 2007 Dez;5(3):184-92.
[Christianson 2005] Christianson DW. Acc Chem Res. 2005 Mrz;38(3):191-201.
[Chung 2015] Chung KK. Methods Mol Biol. 2015 Aug;1292:195-201.
[Cooke 1966] Cooke WT, Smith WT. Brain. 1966 Dez;89(4):683-722.
[Cox 2016] O.H.Cox, R.S.Lee. Medical Epigenetics 2016, Pages 127-146
[Carvalho 2008] Carvalho AL, Caldeira MV, Santos SD, Duarte CB (2008) Role of the brain-derived neurotrophic factor at glutamatergic synapses. Br J Pharmacol 153(1):S310–S324.
[Chandra 2008] Chandra V, Pandav R, Dodge HH, Johnston JM, Belle SH, DeKosky ST, Ganguli M. Neurology. 2001 Sep;57(6):985-9.
[Christianson 2001] Christianson DW. Acc Chem Res. 2005 Mrz;38(3):191-201.
[Cousins 1996] Cousins RJ. Zinc. In: Ziegler EE, Filer LJ, eds. Present Knowledge in Nutrition. Washington DC: ILSI Press; 1996.
[Crapper 1988] Crapper McLachlarf DR, et al. Environ Geochem Health. 1988 Jun;11(2):45-53.
[Crugnola 2010] Crugnola V, et al. Arch Neurol. 2010 Jul;67(7):849-54.
[Cui 2006] Cui X, Zuo P, Zhang Q, Li X, Hu Y, Long J, Packer L, Liu J. J Neurosci Res. 2006 Mrz;83(8):1584-90.
[Dallman 1993] Dallman MF, Strack AM, Akana SF, Bradbury MJ, Hanson ES, Scribner KA, Smith M. Front Neuroendocrinol. 1993 Okt;14(4):303-47.
[Dankner 2005] Dankner R, Chetrit A, Lubin F, Sela BA. Aging Clin Exp Res. 2005 Mrz;16(6):437-42.
[Darmaun 1988] Darmaun D, Matthews DE, Bier DM. Am J Physiol. 1988 Sep;255(3 Pt 1):E366-73.
[Dawson-Hughes 2008] Dawson-Hughes B, Harris SS, Ceglia L. Am J Clin Nutr. 2008 Mrz;87(3):662-5.
[da Costa 2005] da Costa KA, Gaffney CE, Fischer LM, Zeisel SH. Am J Clin Nutr. 2005 Feb;81(2):440-4.
[de Feo 1986] De Feo P, Perriello G, De Cosmo S, Ventura MM, Campbell PJ, Brunetti P, Gerich JE, Bolli GB. Diabetes. 1986 Mai;35(5):563-9.
[de Melo 2017] de Melo LGP, et al. Prog Neuropsychopharmacol Biol Psychiatry. 2017 Jan;78:34-50.
[de Pablos 2006] de Pablos RM, Villarán RF, Argüelles S, Herrera AJ, Venero JL, Ayala A, Cano J, Machado A. J Neurosci. 2006 Mai;26(21):5709-19.
[de Punder 2012] de Punder K, Pruimboom L. Nutrients. 2012 Dez;5(3):771-87.
[Del Campo 2017] Del Campo MT, Romo PE, de la Hoz RE, Villamor JM, Mahíllo-Fernández I. Arch Environ Occup Health. 2017 Jan;72(1):39-44.
[Desbonnet 2008] Desbonnet L, Garrett L, Clarke G, Bienenstock J, Dinan TG. J Psychiatr Res. 2008 Feb;43(2):164-74.
[Desport 2001] Desport JC, Preux PM, Magy L, Boirie Y, Vallat JM, Beaufrère B, Couratier P. Am J Clin Nutr. 2001 Sep;74(3):328-34.
[Desport 1999] Desport JC, Preux PM, Truong TC, Vallat JM, Sautereau D, Couratier P. Neurology. 1999 Sep;53(5):1059-63.
[Dhabhar 1998] Dhabhar FS, McEwen BS. Brain Behav Immun. 1998 Mrz;11(4):286-306.
[Dinubile 2010] Dinubile NA. Phys Sportsmed. 2010 Jul;38(2):71-81.
[Drago 2006] Drago S, et al. Scand J Gastroenterol. 2006 Apr;41(4):408-19.
[Drew 2000] Drew PD, Chavis JA. Brain Res Bull. 2000 Aug;52(5):391-6.
[Duchmann 1999] Duchmann R et al, magazin forschung 1/1999. Mukosales Immunsystem im Darm. S. 47-65
[Dutta 1982] Dutta SK, Bustin MP, Russell RM, Costa BS. Ann Intern Med. 1982 Okt;97(4):549-52.
[Dyckner 1985] Dyckner T, Ek B, Nyhlin H, Wester PO. Acta Med Scand. 1985 Jan;218(1):129-31.
[Ehrlich 2015] Ehrlich SD , University of Maryland Medical Center, Glutamine 8/5/2015.
[Ehrström 2004] Ehrström SM, Kornfeld D, Thuresson J, Rylander E. Am J Obstet Gynecol. 2004 Nov;193(4):1376-81.
[Eid 2008] Eid T, Williamson A, Lee TS, Petroff OA, de Lanerolle NC. Epilepsia. 2008 Mrz;49 Suppl 2:42-52.
[Elders 1925] Elders C. Tropical sprue and pernicious anaemia, aetiology and treatment. Lancet 1925;i:75–7.
[Elizabeth 2010] Elizabeth D. Kirby and Daniela Kaufer. Stress – From Molecules to Behavior. 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 978-3-527-32374-6
[Elsea 2015] Elsea CR, Kneiss JA, Wood LJ. Biol Res Nurs. 2015 Okt;17(5):549-57.
[Emanuele 2009] Emanuele E, Orsi P, Boso M, Broglia D, Brondino N, Barale F, di Nemi SU, Politi P. Neurosci Lett. 2009 Nov;471(3):162-5.
[Endres 1987] Endres S et al. Effects of dietary w-3 fatty acids on the acute phase response during endotoxin fever in humans. J Leuk Biol 42:591, 1987
[Engin 2017] Engin A. Adv Exp Med Biol. 2017 Sep;960:53-80.
[Enns 2007] Enns GM, Berry SA, Berry GT, Rhead WJ, Brusilow SW, Hamosh A. N Engl J Med. 2007 Jun;356(22):2282-92.
[Epel 2018] Epel ES. Hormones (Athens). 2018 Jul;8(1):7-22.
[Erickson 2011] Erickson KI, et al. Proc Natl Acad Sci U S A. 2011 Jan;108(7):3017-22.
[Erridge 2007] Erridge C, Attina T, Spickett CM, Webb DJ. Am J Clin Nutr. 2007 Nov;86(5):1286-92.
[Erridge 2010] Erridge C. Br J Nutr. 2010 Sep;105(1):15-23.
[Evans 2007] Evans KE, Leeds JS, Morley S, Sanders DS. Dig Dis Sci. 2009 Nov;55(10):2999-3004.
[Exner 2000] Exner R, Wessner B, Manhart N, Roth E. Wien Klin Wochenschr. 2000 Jul;112(14):610-6.
[Faggiano 2005] Faggiano A, et al. J Clin Endocrinol Metab. 2005 Dez;90(12):6616-22.
[Fasano 2011] Fasano A. Physiol Rev. 2011 Jan;91(1):151-75.
[Fasano 2011b] Fasano A. Clin Rev Allergy Immunol. 2011 Nov;42(1):71-8.
[Fatemi 2007] Fatemi SH. Prog Neuropsychopharmacol Biol Psychiatry. 2007 Okt;32(3):911, author reply 912-3.
[Ferguson 2013] Ferguson JF, et al. Mol Nutr Food Res. 2013 Mai;58(3):601-13.
[Fidler 2011] Fidler JA, et al. FASEB J. 2011 Dez;25(12):4369-77.
[Filippakis 2018] Filippakis A, et al. Muscle Nerve. 2018 Jul.
[Fine 2012] Fine, Edward J., M.D., Emergency Medicine, July 15, 1992;198-201.
[Fischer 2007] Fischer CP. Exerc Immunol Rev. 2007 Jan;12:6-33.
[Fiszman 2009] Fiszman ML, Ricart KC, Latini A, Rodríguez G, Sica RE. Acta Neurol Scand. 2009 Okt;121(2):120-6.
[Flynn 1997] Flynn NE, Wu G. Am J Physiol. 1997 Mrz;272(3 Pt 1):G474-80.
[Forsmark 2018] Forsmark CE. Curr Treat Options Gastroenterol. 2018 Jul.
[Freeman 2016] Freeman HJ. Ann Gastroenterol. 2016 Apr;29(3):241-2.
[Fukui 1995] Fukui H, et al. J Gastroenterol Hepatol. 1995 Jan;10 Suppl 1:S31-4.
[Gadoth 2015] Gadoth A, Nefussy B, Bleiberg M, Klein T, Artman I, Drory VE. JAMA Neurol. 2015 Jun;72(6):676-81.
[Gaetke 1997] Gaetke LM, McClain CJ, Talwalkar RT, Shedlofsky SI. Am J Physiol. 1997 Jun;272(6 Pt 1):E952-6.
[Galland 1983] Galland L. Nutrition and Candidiasis. at the "Yeast - Human Interaction" Symposium in Birmingham, Alabama, December 10,1983.
[Garibotto 1996] Garibotto G, Russo R, Sofia A, Sala MR, Sabatino C, Moscatelli P, Deferrari G, Tizianello A. Miner Electrolyte Metab. 1996 Jan;22(1-3):58-61.
[Gargiulo Monachelli 2011] Gargiulo Monachelli G, et al. Acta Neurol Scand. 2011 Jan;123(1):60-7.
[Garruto 1989] Garruto RM, Shankar SK, Yanagihara R, Salazar AM, Amyx HL, Gajdusek DC. Acta Neuropathol. 1989 Jan;78(2):210-9.
[Ghanim 2009] Ghanim H, et al. Diabetes Care. 2009 Sep;32(12):2281-7.
[Gilad 1990] Gilad GM, Gilad VH, Wyatt RJ, Tizabi Y. Brain Res. 1990 Aug;525(2):335-8.
[Gimson 2018] Gimson A, Schlosser M, Huntley JD, Marchant NL. BMJ Open. 2018 Mai;8(4):e019399.
[Glasbrenner 1991] Glasbrenner B, Malfertheiner P, Büchler M, Kuhn K, Ditschuneit H. Klin Wochenschr. 1991 Feb; 69(4):168-72
[Gleeson 2008] Gleeson M. J Nutr. 2008 Okt;138(10):2045S-2049S.
[Gourley 2009] Gourley SL, Kedves AT, Olausson P, Taylor JR (2009) Neuropsychopharmacology. 34(3):707–716.
[Griesbach 2004] Griesbach GS, Hovda DA, Molteni R, Wu A, Gomez-Pinilla F. Neuroscience. 2004 Sep;125(1):129-39.
[Groschwitz 2009] Groschwitz KR, Hogan SP. J Allergy Clin Immunol. 2009 Mrz;124(1):3-20; quiz 21-2.
[Gu 2010] Gu Z, Nakamura T, Lipton SA. Mol Neurobiol. 2010 Jun;41(2-3):55-72.
[Guder 1987] Guder WG, Häussinger D, Gerok W. J Clin Chem Clin Biochem. 1987 Aug;25(8):457-66.
[Hack 1996] Hack, V., et al. (1996) FASEBJ. 10, 1219-1226.
[Hadjivassiliou 1996] Hadjivassiliou M, Gibson A, Davies-Jones GA, Lobo AJ, Stephenson TJ, Milford-Ward A. Lancet. 1996 Feb;347(8998):369-71.
[Hadjivassiliou 2002] Hadjivassiliou M, et al. Neurology. 2002 Apr;58(8):1221-6.
[Hadjivassiliou 2002b] Hadjivassiliou M, Grünewald RA, Davies-Jones GA. J Neurol Neurosurg Psychiatry. 2002 Apr;72(5):560-3.
[Hadjivassiliou 2003] Hadjivassiliou M, Davies-Jones GA, Sanders DS, Grünewald RA. J Neurol Neurosurg Psychiatry. 2003 Aug;74(9):1221-4.
[Häussinger 1993] Häussinger D, Roth E, Lang F, Gerok W. Lancet. 1993 Mai;341(8856):1330-2.
[Hayton 1995] Hayton, Bruce A., et al, American Journal of Hematology, 1995;48:45-47.
[He 2010] He Y, et al. J Biol Chem. 2010 Jan;285(13):9516-24.
[Heales 2002] Heales SJ, Bolaños JP. Neurochem Int. 2002 Mai;40(6):469-74.
[Helms 2015] Helms ER, Zinn C, Rowlands DS, Naidoo R, Cronin J. Int J Sport Nutr Exerc Metab. 2015 Apr;25(2):163-70.
[Henrotte 1986] Henrotte JG. Magnesium. 1986 Jan;5(3-4):201-10.
[Hickson 1995] Hickson RC, Czerwinski SM, Wegrzyn LE. Am J Physiol. 1995 Apr;268(4 Pt 1):E730-4.
[Hickson 1997] Hickson RC, Oehler DT, Byerly RJ, Unterman TG. Proc Soc Exp Biol Med. 1997 Okt;216(1):65-71.
[Hollander 2000] Hollander D. Curr Gastroenterol Rep. 2000 Sep;1(5):410-6.
[Holman 1982] Holman RT, Johnson SB, Hatch TF. Am J Clin Nutr. 1982 Mrz;35(3):617-23.
[Holmes 1997] Holmes GKT. Neurological and psychiatric complications in coeliac disease. In: Gobbi G, Anderman F, Naccarato S, et al, eds. Epilepsy and other neurological disorders in coeliac disease. London: John Libbey, 1997
[Homocysteine Lowering Trialists' Collaboration 1998] Homocysteine Lowering Trialists' Collaboration. BMJ. 1998 Mrz;316(7135):894-8.
[Hu 2016] Hu X, Wang T, Jin F. Sci China Life Sci. 2016 Jun;59(10):1006-1023.
[Hu 2017] Hu Y, Cao C, Qin XY, Yu Y, Yuan J, Zhao Y, Cheng Y. Sci Rep. 2017 Aug;7(1):9094.
[Ignatowski 1996] Ignatowski TA, Gallant S, Spengler RN. J Neuroimmunol. 1996 Apr;65(2):107-17.
[Iovino 1998] Iovino P, Ciacci C, Sabbatini F, Acioli DM, D'Argenio G, Mazzacca G. Am J Gastroenterol. 1998 Aug;93(8):1243-9.
[Isolauri 1992] Isolauri, Erika, M.D., et al, The Journal of Pediatrics, January 1992;120(1):9-15.
[Iwasaki 1992] Iwasaki Y, Ikeda K, Kinoshita M. J Neurol Sci. 1992 Feb;107(2):219-22.
[Iwashita 2005] Iwashita S, et al. J Appl Physiol (1985). 2005 Jul;99(5):1858-65.
[Jackson 2011] Jackson JR, Eaton WW, Cascella NG, Fasano A, Kelly DL. Psychiatr Q. 2011 Aug;83(1):91-102.
[Jacobs 1996] Jacobs D. (Ed.) Laboratory test handbook. 4th ed. Lexi-Corp., Hudson, OH; 1996: 112–114
[Jaffe 1992] Jaffe, Russell, M.D. and Kruesi, Oscar, International Clinical Nutrition Review, January 1992;12(1):9-26.
[Jaspan 1992] Jaspan JB, Wollman RL, Bernstein L, Rubenstein AH. Medicine (Baltimore). 1982 Jan;61(1):33-44.
[Jianmin 2015] Jianmin Chen PhD, Karl Herrup PhD, in Diet and Nutrition in Dementia and Cognitive Decline, 2015
[Jawhara 2008] Jawhara S, et al, Poulain D. J Infect Dis. 2008 Apr;197(7):972-80.
[Jensen 2008] Jensen GL. JPEN J Parenter Enteral Nutr. 2008 Nov;32(6):656-9.
[Jurnak 2015] Jurnak F. Nutr Metab Insights. 2015 Dez;8(Suppl 1):57-77.
[Juuti-Uusitalo 2007] Juuti-Uusitalo K, Mäki M, Kainulainen H, Isola J, Kaukinen K. Clin Exp Immunol. 2007 Sep;150(2):294-305.
[Kalach 2001] Kalach N, Rocchiccioli F, de Boissieu D, Benhamou P-H, Dupont C, Acta Paediatr, 2001;90:499-504.
[Kan 2015] Kan MJ, et al. J Neurosci. 2015 Apr;35(15):5969-82.
[Kanofsky 1991] Kanofsky JD, Sandyk R. Int J Neurosci. 1991 Nov;61(1-2):87-90.
[Kardalas 2018] Kardalas E, Paschou SA, Anagnostis P, Muscogiuri G, Siasos G, Vryonidou A. Endocr Connect. 2018 Apr;7(4):R135-R146.
[Kelly 2003] Kelly D, Wischmeyer PE. Curr Opin Clin Nutr Metab Care. 2003 Feb;6(2):217-22.
[Kanofsky 1991] Kanofsky JD, Sandyk R. Int J Neurosci. 1991 Nov;61(1-2):87-90.
[Kang 2005] Kang YJ, Zhou Z. Mol Aspects Med. 2005 Aug;26(4-5):391-404.
[Kerr 1993] Kerr D, Sherwin RS, Pavalkis F, Fayad PB, Sikorski L, Rife F, Tamborlane WV, During MJ. Ann Intern Med. 1993 Okt;119(8):799-804.
[Kiecolt-Glaser 2010] Kiecolt-Glaser JK. Psychosom Med. 2010 Apr;72(4):365-9.
[Kinscherf 1996] Kinscherf R, et al. J Mol Med (Berl). 1996 Jul;74(7):393-400.
[Kim 2008] Kim CJ, Kovacs-Nolan J, Yang C, Archbold T, Fan MZ, Mine Y. Biochim Biophys Acta. 2008 Okt;1790(10):1161-9.
[Kinscherf 1996] Kinscherf R, et al. J Mol Med (Berl). 1996 Jul;74(7):393-400.
[Knowles 2007] Knowles SR, Nelson EA, Palombo EA. Biol Psychol. 2007 Feb;77(2):132-7.
[Köhler 2016] Köhler CA, Maes M, Slyepchenko A, Berk M, Solmi M, Lanctôt KL, Carvalho AF. Curr Pharm Des. 2016 Jul;22(40):6152-6166.
[Koelsch 2016] Koelsch S, Boehlig A, Hohenadel M, Nitsche I, Bauer K, Sack U. Sci Rep. 2016 Mrz;6:23008.
[Kompan 2001] Kompan L, Kompan D, Eur J Surg, 2001;167:570-574.
[Kruman 2000] Kruman II, Culmsee C, Chan SL, Kruman Y, Guo Z, Penix L, Mattson MP. J Neurosci. 2000 Sep;20(18):6920-6.
[Löffler 2006] Löffler G, Petro E. Petrides, Peter C. Heinrich: Biochemie & Pathobiochemie. Springer Medizin Verlag, Heidelberg 2006, ISBN 3-540-32680-4, S. 1040.
[Kuhn 2001] Kuhn KS, Schuhmann K, Stehle P, Darmaun D, Fürst P. Am J Clin Nutr. 1999 Sep;70(4):484-9.
[Kukkonen-Harjula 1989] Kukkonen-Harjula K, et al. Eur J Appl Physiol Occup Physiol. 1989 Jan;58(5):543-50.
[Kula 2015] Kula J, Blasiak A, Czerw A, Tylko G, Sowa J, Hess G. Pflugers Arch. 2015 Jun;468(4):679-91.
[Kumamoto 2011] Kumamoto CA. Curr Opin Microbiol. 2011 Jul;14(4):386-91.
[Kumar 2017] Kumar P, Raman T, Swain MM, Mishra R, Pal A. Mol Neurobiol. 2017 Jan;54(1):238-254.
[Lacey 1990] Lacey JM, Wilmore DW. Nutr Rev. 1990 Aug;48(8):297-309.
[Lakier 2003] Lakier Smith L. Sports Med. 2003 Aug;33(5):347-64.
[Lammers 2007] Lammers KM, et al. Gastroenterology. 2007 Jul;135(1):194-204.e3.
[Lands 1999] Lands LC, Grey VL, Smountas AA. J Appl Physiol (1985). 1999 Okt;87(4):1381-5.
[Lauretti 2016] Lauretti E, Li JG, Di Meco A, Praticò D. Transl Psychiatry. 2016 Okt;7(1):e1020.
[Lebenthal 2002] Lebenthal E, Branski D. J Pediatr Gastroenterol Nutr. 2002 Okt;35(4):472-4.
[Leblhuber 2014] Leblhuber F, Geisler S, Steiner K, Fuchs D, Schütz B. J Neural Transm (Vienna). 2014 Nov;122(9):1319-22.
[Leclercq 2014] Leclercq S, et al. Proc Natl Acad Sci U S A. 2014 Okt;111(42):E4485-93.
[Leclercq 2016] Leclercq S, Forsythe P, Bienenstock J. Can J Psychiatry. 2016 Jun;61(4):204-13.
[Lee 2005] Lee SK, Green PH. Curr Opin Rheumatol. 2005 Dez;18(1):101-7.
[Lee 2015] Lee CC, Perkins BA, Kayaniyil S, Harris SB, Retnakaran R, Gerstein HC, Zinman B, Hanley AJ. Diabetes Care. 2015 Mai;38(5):793-800.
[Lehman 2012] Lehman EJ, Hein MJ, Baron SL, Gersic CM. Neurology. 2012 Sep;79(19):1970-4.
[Levens 1981] Levens NR, Peach MJ, Carey RM. J Clin Invest. 1981 Apr; 67(4):1197-207.
[Lerner 2002] Lerner V, Miodownik C, Kaptsan A, Cohen H, Loewenthal U, Kotler M. J Clin Psychiatry. 2002 Feb;63(1):54-8.
[Li 1994] Li J, Langkamp-Henken B, Suzuki K, Stahlgren LH. JPEN J Parenter Enteral Nutr. 1994 Jul;18(4):303-7.
[Lin 2011] Lin XL, Li Z, Zuo XL. Zhonghua Fu Chan Ke Za Zhi. 2011 Nov;46(7):496-500.
[Linde 1981] Linde R, Stone W, Kirshner H. JAMA. 1981 Okt;246(17):1899-900.
[Lindenbaum 1994] Lindenbaum J, Rosenberg IH, Wilson PW, Stabler SP, Allen RH. Am J Clin Nutr. 1994 Jul;60(1):2-11.
[Liu 2000] Liu D, Ling X, Wen J, Liu J. J Neurochem. 2000 Nov;75(5):2144-54.
[Liu 2004] Liu Q, Duan ZP, Ha DK, Bengmark S, Kurtovic J, Riordan SM. Hepatology. 2004 Mai;39(5):1441-9.
[Lu 2003] Lu B (2003) BDNF and activity-dependent synaptic modulation. Learn Mem 10:86–98.
[Lucassen 2008] Lucassen PJ, et al. Eur Neuropsychopharmacol. 2008 Nov;20(1):1-17.
[Lucchetti 2008] Lucchetti G, Oliveira AB, Mercante JP, Peres MF. Curr Pain Headache Rep. 2012 Okt;16(5):399-406.
[Lüningschrör 2016] Lüningschrör P, et al. Nat Commun. 2016 Sep;8(1):678.
[Lundsgaard 1996] Lundsgaard C, Hamberg O, Thomsen OO, Nielsen OH, Vilstrup H. J Hepatol. 1996 Mai;24(5):587-93.
[Brittenden 1994] Brittenden J, Heys SD, Ross J, Park KG, Eremin O. Clin Sci (Lond). 1994 Feb;86(2):123-32.
[Maciejewski 2006] Maciejewski PK, Rothman DL. Neurochem Int. 2006 Dez;52(4-5):809-25.
[Machts 2018] Machts J, Vielhaber S, Kollewe K, Petri S, Kaufmann J, Schoenfeld MA. Front Neurol. 2018 Sep;9:565.
[Madeddu 2012] Madeddu C, et al. Clin Nutr. 2012 Apr;31(2):176-82.
[Maes 2007] Maes M, Mihaylova I, Leunis JC. Neuro Endocrinol Lett. 2007 Okt;28(6):861-7.
[Maier 2015] Maier SU, Makwana AB, Hare TA. Neuron. 2015 Feb;87(3):621-31.
[Mainardi 2002] Mainardi E, Montanelli A, Dotti M, Nano R, Moscato G. J Clin Gastroenterol. 2002 Aug;35(3):245-8.
[Maldonado 2002] Maldonado ME, Williams RC, Adair JC, Hart BL, Gregg L, Sibbitt WL. J Rheumatol. 2002 Mrz;29(3):633-5.
[Marin 2011] Marin B, Desport JC, Kajeu P, Jesus P, Nicolaud B, Nicol M, Preux PM, Couratier P. J Neurol Neurosurg Psychiatry. 2011 Jun;82(6):628-34.
[Mariosa 2015] Mariosa D, Kamel F, Bellocco R, Ye W, Fang F. Eur J Neurol. 2015 Nov;22(11):1436-42.
[Mathurin 2000] Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Hepatology. 2000 Okt;32(5):1008-17.
[May 1986] May RC, Kelly RA, Mitch WE. J Clin Invest. 1986 Feb;77(2):614-21.
[McCance 1934] McCance RA, Sheldon W, Widdowson EM. Arch Dis Child. 1934 Aug;9(52):251-8.
[McCance 2014] McCrea CE, et al. J Transl Med. 2014 Sep;13:7.
[McGeer 2002] McGeer PL, McGeer EG. Muscle Nerve. 2002 Oct; 26(4):459-70.
[McGough 2016] McGough G. Nurs Stand. 2016 Jun;25(51):30.
[McRae 2013] McRae MP. J Chiropr Med. 2013 Mrz;12(1):20-5.
[Meldrum 2000] Meldrum BS. J Nutr. 2000 Mrz;130(4S Suppl):1007S-15S.
[Meeusen 2013] Meeusen R, et al, European College of Sport Science, American College of Sports Medicine. Med Sci Sports Exerc. 2013 Jan;45(1):186-205.
[Merry 2016] Merry TL, Ristow M. J Physiol. 2016 Sep;594(18):5135-47.
[Ristow 2009] Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Blüher M. Proc Natl Acad Sci U S A. 2009 Mai;106(21):8665-70.
[Miller 1999] Miller AL. Altern Med Rev. 1999 Sep;4(4):239-48.
[Miller 2002] Miller GE, Cohen S, Ritchey AK. Health Psychol. 2002 Nov;21(6):531-41.
[Miller 2008] Miller AH, Maletic V, Raison CL. Biol Psychiatry. 2008 Okt;65(9):732-41.
[Miller 2010] Miller KC, et al. Med Sci Sports Exerc. 2010 Mai;42(5):953-61.
[Miller 2018] Miller KC, Mack G, Knight KL. J Athl Train. 2018 Aug;44(5):454-61.
[Mirescu 2006] Mirescu C, Peters JD, Noiman L, Gould E. Proc Natl Acad Sci U S A. 2006 Dez;103(50):19170-5.
[Mittendorfer 2001] Mittendorfer B, Volpi E, Wolfe RR. Am J Physiol Endocrinol Metab. 2001 Feb;280(2):E323-33.
[Moat 2000] Moat SJ, Bonham JR, Cragg RA, Powers HJ. Free Radic Res. 2000 Feb;32(2):171-9.
[Molteni 2002] Molteni R, Barnard RJ, Ying Z, Roberts CK, Gómez-Pinilla F. Neuroscience. 2002 Jun;112(4):803-14.
[Mytilineou 2002] Mytilineou C, Kramer BC, Yabut JA. Parkinsonism Relat Disord. 2002 Sep;8(6):385-7.
[Nakamura 2007] Nakamura T, Gu Z, Lipton SA. Aging Cell. 2007 Jun;6(3):351-9.
[Nienaber 2016] Nienaber A, Dolman RC, van Graan AE, Blaauw R. Nutr J. 2016 Mrz;15(1):73.
[Nieves 2018] Nieves JW, et al. JAMA Neurol. 2018 Jul;73(12):1425-1432.
[Newsholme 1989] Newsholme EA, Newsholme P, Curi R, Crabtree B, Ardawi MSM (1989) Glutamine metabolism in different tissues – its physiological and pathological importance. In: Kinney JM, Borum PR (eds) Perspectives in Clinical Nutrition. Urban & Schwarzenberg, Baltimore- München, pp 71– 98
[Neu 2002] Neu J, DeMarco V, Li N. Curr Opin Clin Nutr Metab Care. 2002 Jan;5(1):69-75.
[Newsholme 2001] Newsholme P. J Nutr. 2001 Sep;131(9 Suppl):2515S-22S; discussion 2523S-4S.
[Newsholme 1986] Newsholme P, Curi R, Gordon S, Newsholme EA. Biochem J. 1986 Okt;239(1):121-5.
[Nguyen 2017] Nguyen B, Ding D, Mihrshahi S. BMJ Open. 2017 Mrz;7(3):e014201.
[Nishitani 2005] Nishitani N, Sakakibara H. Int J Obes (Lond). 2005 Okt;30(3):528-33.
[Nohmi 1994] Nohmi T, Abe S, Tansho S, Yamaguchi H. Microbiol Immunol. 1994 Jan;38(12):977-82.
[Nose 2009] Nose K, Yang H, Sun X, Nose S, Koga H, Feng Y, Miyasaka E, Teitelbaum DH. J Interferon Cytokine Res. 2009 Dez;30(2):67-80.
[O'SULLIVAN 1958] O'SULLIVAN JB, DYNES JB, MURPHY R. Lahey Clin Bull. 1958 Jan;10(7):218-20.
[Owen 1963] OWEN EE, ROBINSON RR. J Clin Invest. 1963 Feb;42:263-76.
[Palop 2006] Palop JJ, Chin J, Mucke L. Nature. 2006 Okt;443(7113):768-73.
[Petrou 2011] Petrou M, Pop-Busui R, Foerster BR, Edden RA, Callaghan BC, Harte SE, Harris RE, Clauw DJ, Feldman EL. Acad Radiol. 2011 Dez;19(5):607-12.
[O'Dwyer 1989] O'Dwyer ST, Smith RJ, Hwang TL, Wilmore DW. JPEN J Parenter Enteral Nutr. 1989 Nov;13(6):579-85.
[O'Mahony 2010] O'Mahony SM, Hyland NP, Dinan TG, Cryan JF. Psychopharmacology (Berl). 2010 Apr;214(1):71-88.
[Parlesak 2000] Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C (2000). J. Hepatol. 32 (5): 742–7.
[Parekh 2015] Parekh B. F1000Res. 2015 Sep;4:119.
[Patacchioli 2004] Patacchioli FR, et al. J Endocrinol Invest. 2004 Apr;26(12):RC23-5.
[Pendyala 2011] Pendyala S, Walker JM, Holt PR. Gastroenterology. 2011 Okt;142(5):1100-1101.e2.
[Petty 2011] Petty F, Kramer GL, Gullion CM, Rush AJ. Biol Psychiatry. 1992 Aug;32(4):354-63.
[Pocernich 1992] Pocernich CB, Butterfield DA. Biochim Biophys Acta. 2011 Aug;1822(5):625-30.
[Pomara 1992] Pomara N, Singh R, Deptula D, Chou JC, Schwartz MB, LeWitt PA. Am J Psychiatry. 1992 Feb;149(2):251-4.
[Prado 2017] Prado LGR, et al. Einstein (Sao Paulo). 2017 Aug;15(1):58-60.
[Palmer 2007] Palmer K, Berger AK, Monastero R, Winblad B, Bäckman L, Fratiglioni L. Neurology. 2007 Mai;68(19):1596-602.
[Prinz 2017] Mass E, et al. Nature. 2017 Mrz.
[Purohit 2007] Purohit V, et al. Alcohol. 2007 Dez;42(5):349-61.
[Qin 2007] Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Glia. 2007 Jan;55(5):453-62.
[Rahman 2000] Rahman I, MacNee W. Eur Respir J. 2000 Okt;16(3):534-54.
[Rahman 2000] Rama Rao KV, Jayakumar AR, Norenberg DM. Metab Brain Dis. 2003 Jun;18(2):113-27.
[Ramasamy 2006] Ramasamy S, et al. Am J Physiol Gastrointest Liver Physiol. 2006 Feb;291(2):G288-96.
[Rao 1991] Rao VL, Murthy CR. Neurosci Lett. 1991 Mai;126(1):13-7.
[Rao 2004] Rao TV, Tharakan JK, Jacob PC. Clin Neuropathol. 2004 Jun;23(3):99-101.
[Rendeiro 2012] Rendeiro C, Vauzour D, Rattray M, Waffo-Téguo P, Mérillon JM, Butler LT, Williams CM, Spencer JP. PLoS One. 2012 Nov;8(5):e63535.
[Rennie 2001] Rennie MJ, Bowtell JL, Bruce M, Khogali SE. J Nutr. 2001 Sep;131(9 Suppl):2488S-90S; discussion 2496S-7S.
[Richardson 1976] Richardson CT, Walsh JH, Hicks MI, Fordtran JS. J Clin Invest. 1976 Sep;58(3):623-31.
[Ridel 2004] Ridel KR, Leslie ND, Gilbert DL. Pediatr Neurol. 2004 Nov;33(3):153-61.
[Rivest 2000] Rivest S, Lacroix S, Vallières L, Nadeau S, Zhang J, Laflamme N. Proc Soc Exp Biol Med. 2000 Jan;223(1):22-38.
[Rizza 1982] Rizza RA, Mandarino LJ, Gerich JE. J Clin Endocrinol Metab. 1982 Jan;54(1):131-8.
[Roberts 2013] Roberts CJ, Campbell IC, Troop N. Eur Eat Disord Rev. 2013 Apr;22(1):77-82.
[Roselle 1982] Roselle GA, Mendenhall CL. J Clin Lab Immunol. 1982 Okt;9(1):33-7.
[Rosenberg 1992] Rosenberg IH, Miller JW. Am J Clin Nutr. 1992 Jun;55(6 Suppl):1237S-1243S.
[Roth 1982] Roth E, Funovics J, Mühlbacher F, Schemper M, Mauritz W, Sporn P, Fritsch A. Clin Nutr. 1982 Mrz;1(1):25-41.
[Roth 1990] Roth E, Karner J, Ollenschlager G. JPEN J Parenter Enteral Nutr. 1990 Jul;14(4 Suppl):130S-136S.
[Roth 2004] Roth E, Shock, 2004;21(Suppl):123(Abstract 489).
[Roth 2008] Roth E. J Nutr. 2008 Okt;138(10):2025S-2031S.
[Sadeghi 1999] Sadeghi S, Wallace FA, Calder PC. Immunology. 1999 Mai;96(3):404-10.
[Sategna-Guidetti 2001] Sategna-Guidetti C, Volta U, Ciacci C, Usai P, Carlino A, De Franceschi L, Camera A, Pelli A, Brossa C. Am J Gastroenterol. 2001 Mrz;96(3):751-7.
[Schaaf 2002] Schaaf MJ, De Kloet ER, Vreugdenhil E. Stress. 2000 Mai;3(3):201-8.
[Schulze 2009] Schulze J, Sonnenborn U. Dtsch Arztebl Int. 2009 Apr;106(51-52):837-42.
[Schwartz 1991] Schwartz, R.H. IgE-mediated allergic reactions to cow's milk. Immunol Allergy Clin N Amer. 1991; 11: 717-742.
[Schwarz 1996] Schwarz S, Husstedt I, Bertram HP, Kuchelmeister K. J Neurol Neurosurg Psychiatry. 1996 Jun;60(6):698.
[Seeram 2001] Seeram NP, Momin RA, Nair MG, Bourquin LD. Phytomedicine. 2001 Nov;8(5):362-9.
[Seidman 1991] Seidman E, et al. J Pediatr Gastroenterol Nutr. 1991 Mai;12(4):424-38.
[Seifert 2006] Seifert G, Schilling K, Steinhäuser C. Nat Rev Neurosci. 2006 Mrz;7(3):194-206.
[Seitz 1998] Seitz HK, Oneta CM. Nutr Rev. 1998 Feb; 56(2 Pt 1):52-60.
[Sevinc 2016] Sevinc E, Sevinc N, Akar HH, Ozelcoskun BD, Sezgin GC, Arslan D, Kendirci M. Asia Pac J Clin Nutr. 2016 Jul;25(3):452-6.
[Shahani 2011] Shahani N, Sawa A. Antioxid Redox Signal. 2011 Apr;14(8):1493-504.
[Sharma 1994] Sharma SK, Bolster B, Dakshinamurti K. J Neurol Sci. 1994 Jan;121(1):1-9.
[Sharma 2015] Sharma ST, Nieman LK, Feelders RA. Clin Epidemiol. 2015 Sep;7:281-93.
[Sheldon 2007] Sheldon AL, Robinson MB. Neurochem Int. 2007 Feb;51(6-7):333-55.
[Sherry 1993] Sherry A. Rogers, M.D.; Tötet Sie Ihr Kardiologe? Journal für Orthomolekulare Medizin 2/93
[Shibata 1995] Shibata S, Tominaga K, Watanabe S. Eur J Pharmacol. 1995 Jan;273(1-2):191-5.
[Shimmura 2011] Shimmura C, et al. PLoS One. 2011 Mrz;6(10):e25340.
[Simon 2013] Simon NG, Kiernan MC. J Neurol. 2013 Jul;260(7):1743-7.
[Sjögren 1988] Sjögren A, Florén CH, Nilsson A. Scand J Gastroenterol. 1988 Jun;23(5):555-61.
[Smeyne 2012] Smeyne M, Smeyne RJ. Free Radic Biol Med. 2012 Sep;62:13-25.
[Smith 1990] Smith RJ. JPEN J Parenter Enteral Nutr. 1990 Jul;14(4 Suppl):40S-44S.
[Smith 2000] Smith LL. Med Sci Sports Exerc. 2000 Feb;32(2):317-31.
[Solomon 2005] Solomon LR. Blood. 2005 Feb;105(3):978-85; author reply 1137.
[Souba 1990] Souba WW, Herskowitz K, Klimberg VS, Salloum RM, Plumley DA, Flynn TC, Copeland EM. Ann Surg. 1990 Mai;211(5):543-9; discussion 549-51.
[Soreq 2009] Hermona Soreq, Alon Friedman, and Daniela Kaufer. Stress – From Molecules to Behavior. November 2009, Wiley-Blackwell, Weinheim ISBN: 978-3-527-32374-6
[Smythies 1996] Smythies JR. J R Soc Med. 1996 Mai;89(5):241.
[Spataro 2015] Spataro R, Volanti P, Vitale F, Meli F, Colletti T, Di Natale A, La Bella V. J Neurol Sci. 2015 Nov;358(1-2):282-6.
[Spodenkiewicz 2016] Spodenkiewicz M, Diez-Fernandez C, Rüfenacht V, Gemperle-Britschgi C, Häberle J. Biology (Basel). 2016 Jul;5(4).
[Stöckler-Ipsiroglu 1997] Stöckler-Ipsiroglu S. J Pediatr. 1997 Okt;131(4):510-1.
[Stone 2016] Stone MS, Martyn L, Weaver CM. Nutrients. 2016 Jul;8(7).
[Streeter 2007] Streeter CC, Jensen JE, Perlmutter RM, Cabral HJ, Tian H, Terhune DB, Ciraulo DA, Renshaw PF. J Altern Complement Med. 2007 Mai;13(4):419-26.
[Stumvoll 1999] Stumvoll M, Perriello G, Meyer C, Gerich J. Kidney Int. 1999 Feb;55(3):778-92.
[Suomalainen 1992] Suomalainen H, Isolauri E, Kaila M, Virtanen E, Arvilommi H. Gut. 1992 Sep;33(9):1179-83.
[Suzuki 2009] Suzuki T, Hara H. J Nutr. 2009 Mrz;139(5):965-74.
[Taibi 2004] Taibi DM, Bourguignon C, et al, Holist Nurs Pract, May/June 2004;18(3):120-126.
[Taylor 2011] Taylor L, Curthoys NP. Biochem Mol Biol Educ. 2011 Jun;32(5):291-304.
[Tawakol 2016] Tawakol A, et al. Lancet. 2016 Jun;389(10071):834-845.
[Terzolo 2004] Terzolo M, et al. J Clin Endocrinol Metab. 2004 Aug;89(8):3745-51.
[Tesfaye 2004] Tesfaye N, Seaquist ER. Diabetes Care. 2004 Feb;27(2):628-9.
[The DCCT Research Group 1991] The DCCT Research Group, Am J Med, 1991; 90:450–59.
[Thorne 2007] Thorne Research, Inc. (2007). Silybum marianum (milk thistle) [monograph]. Alter native Medicine Review, 4(4), 272-274. Retrieved August 7, 2007
[Tohgi 1999] Tohgi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C. Ann Neurol. 1999 Jul;46(1):129-31.
[Tomlinson 2007] Tomlinson DR, Gardiner NJ. Nat Rev Neurosci. 2007 Dez;9(1):36-45.
[Townsend 2003] Townsend DM, Tew KD, Tapiero H. Biomed Pharmacother. 2003 Jun;57(3-4):145-55.
[Travison 2007] Travison TG, O'Donnell AB, Araujo AB, Matsumoto AM, McKinlay JB. Clin Endocrinol. 2007 Jul;67(1):71-7.
[Turner 2013] Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ. Neurology. 2013 Aug;81(14):1222-5.
[Turner 2016] Turner MR, Goldacre R, Talbot K, Goldacre MJ. Ann Neurol. 2016 Dez;80(6):935-938.
[Uehara 2007] Uehara T. Antioxid Redox Signal. 2007 Mai;9(5):597-601.
[Unknown 1967] Lancet. 1967 Mrz;1(7491):664-5.
[Vahdat 2001] Vahdat L, Papadopoulos K, Lange D, et al. Reduction of paclitaxel-induced peripheral neuropathy with glutamine. Clin Cancer Res 2001;7:1192–1197.
[Van Way 1991] Van Way CW. Surg Clin North Am. 1991 Jun;71(3):537-48.
[Vahdat 1993] Vamier, M. et al., Clinical Nutrition (1993) Volume 12, Supplement 2, Page 1
[Vanuytsel 2013] Vanuytsel T, et al. Gut. 2013 Okt;63(8):1293-9.
[Velissaris 2017] Velissaris D, et al, Ellul J. J Clin Med Res. 2017 Jun;9(6):461-465.
[Veltri 2015] Veltri KT, Mason C. P T. 2015 Mrz;40(3):185-90.
[Ventura 1999] Ventura A, Magazzù G, Greco L. Gastroenterology. 1999 Jul;117(2):297-303.
[Vicennati 2000] Vicennati V, Pasquali R. J Clin Endocrinol Metab. 2000 Nov;85(11):4093-8.
[Visser 2017] Visser AE, et al. J Neurol. 2017 Apr;264(4):694-700.
[Waggoner 1999] Waggoner DJ, Bartnikas TB, Gitlin JD. Neurobiol Dis. 1999 Aug;6(4):221-30.
[Wald 1982] Wald A, Adibi SA. Am J Physiol. 1982 Feb;242(2):G85-8.
[Wang 1996] Wang MD, Little J, Gomes J, Cashman NR, Krewski D. Neurotoxicology. 2016 Jun;61:101-130.
[Walsh 2017] Walsh EI, Shaw M, Sachdev P, Anstey KJ, Cherbuin N. Diabetes Metab. 2017 Mai.
[Wang 2007] Wang WS, et al. Oncologist. 2007 Apr;12(3):312-9.
[Wang 2015] Wang WY, Tan MS, Yu JT, Tan L. Ann Transl Med. 2015 Mrz;3(10):136.
[Wang 2016] Wang MD, Little J, Gomes J, Cashman NR, Krewski D. Neurotoxicology. 2016 Jun;61:101-130.
[Wallace 2005] Wallace JL, Devchand PR. Br J Pharmacol. 2005 Mrz;145(3):275-82.
[Wedler 1984] Wedler FC, Denman RB. Curr Top Cell Regul. 1984 Jan;24:153-69.
[Welbourne 1995] Welbourne TC. Am J Clin Nutr. 1995 Mai;61(5):1058-61.
[Welbourne 1987] Welbourne TC. Am J Physiol. 1987 Dez;253(6 Pt 2):F1069-76.
[Williams 1991] Williams B, Layward E, Walls J. Clin Sci (Lond). 1991 Mai;80(5):457-62.
[Wills 2018] Wills AM, et al. MDA Clinical Research Network. Lancet. 2018 Jul;383(9934):2065-2072.
[Wu 2006] Wu A, Ying Z, Gomez-Pinilla F. Exp Neurol. 2006 Feb;197(2):309-17.
[Wu 2004] Wu A, Ying Z, Gomez-Pinilla F. Eur J Neurosci. 2004 Apr;19(7):1699-707.
[Yager 1999] Yager J, et al, Am J Psychiatry, September 1999;156(9):1432-1438.
[Yamaguchi 2006] Yamaguchi N, Sugita R, Miki A, Takemura N, Kawabata J, Watanabe J, Sonoyama K. Gut. 2006 Jan;55(7):954-60.
[Yasui 1997] Yasui M, Ota K, Yoshida M. Magnes Res. 1997 Mrz;10(1):39-50.
[Yeaman 2001] Yeaman SJ, Armstrong JL, Bonavaud SM, Poinasamy D, Pickersgill L, Halse R. Biochem Soc Trans. 2001 Aug;29(Pt 4):537-41.
[Yehuda 1998] Yehuda R, Siever LJ, Teicher MH, Levengood RA, Gerber DK, Schmeidler J, Yang RK. Biol Psychiatry. 1998 Jul;44(1):56-63.
[Yemma 1994] Yemma JJ, Berk MP. Cytobios. 1994 Jan;77(310):147-58.
[Yudkin 1978] Yudkin J. Lipids. 1978 Mai;13(5):370-2.
[Zhang 2008] Zhang R, Miller RG, Gascon R, Champion S, Katz J, Lancero M, Narvaez A, Honrada R, Ruvalcaba D, McGrath MS. J Neuroimmunol. 2008 Mai;206(1-2):121-4.
[Zhong 2010] Zhong W, McClain CJ, Cave M, Kang YJ, Zhou Z. Am J Physiol Gastrointest Liver Physiol. 2010 Feb;298(5):G625-33.
[Zhou 2003] Zhou YP, Jiang ZM, Sun YH, Wang XR, Ma EL, Wilmore D. JPEN J Parenter Enteral Nutr. 2003 Aug;27(4):241-5.
[Zipser 2003] Zipser RD, Patel S, Yahya KZ, Baisch DW, Monarch E. Dig Dis Sci. 2003 Mai;48(4):761-4.
